
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Flexible Protein Simulations of Monoclonal Antibodies to Enable Biotherapeutic Development
Summary
All-atom modeling and dynamics refines design of protein therapeutics by incorporating dynamics in low and high concentration formulations. Molecular dynamics simulations of the NISTmAb monoclonal antibody reference material, a 130 kDa protein that is a standard reference material widely used in the biotechnology industry, provides insight into its function as well as tangible data for use in a variety of biophysical applications. In several projects, computational work integrates with experimental work to tell us something about the critical quality attributes of this highly flexible molecule.
Goal: Incorporate all-atom dynamics into the biotherapeutic pipeline
1. Grow from the “static sphere” model to interpret experiments
2. Quantify dynamics for mAbs
Description
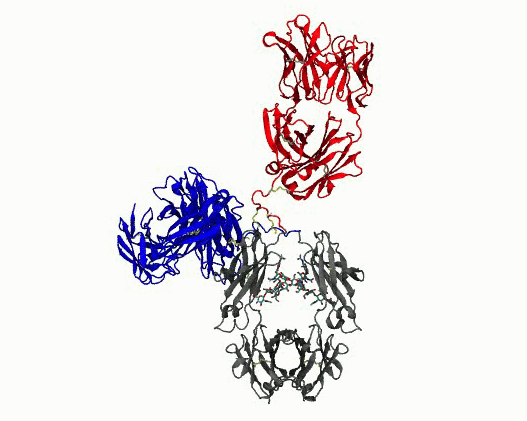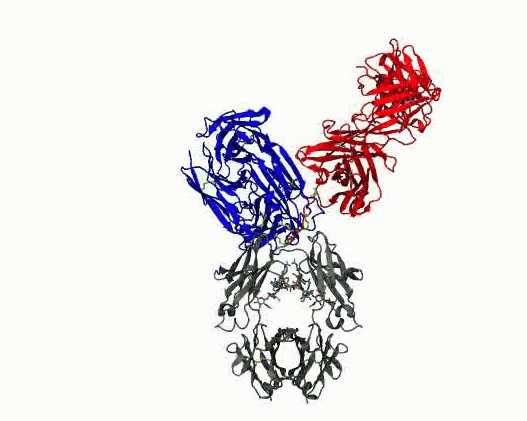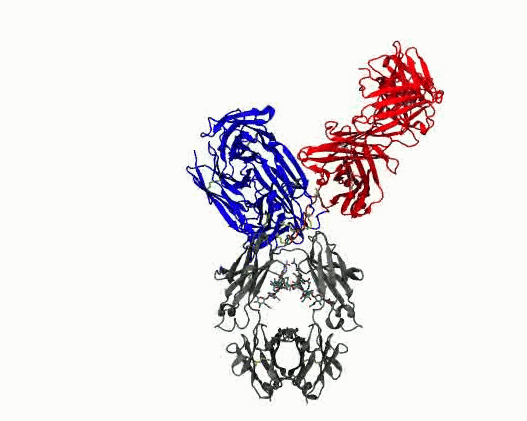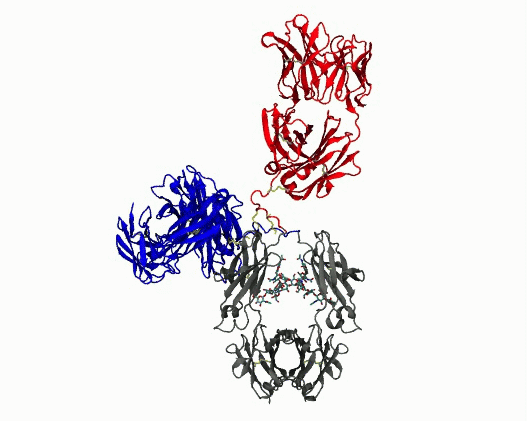
Dynamic motions of the NISTmAb antibody
Grow from the “static sphere” model to interpret experiments. Monoclonal antibodies (mAbs) are important therapeutics. Formulating them at high concentrations is difficult because they tend to cluster and aggregate, which raises viscosity and shortens shelf‑life. A major, yet poorly understood, contributor to this behavior is the flexibility of the hinge region that connects the Fab and Fc domains. Because the hinge is highly flexible and largely unstructured, conventional structural experiments (cryo‑EM, X‑ray crystallography) provide little insight. Experimental data have historically been interpreted using structure models that reduce the complexity to a static sphere. The work we are pursuing clarifies the molecular determinants of hinge flexibility, providing a mechanistic basis for how hinge architecture affects antibody packing, clustering, and aggregation in concentrated formulations. The results will benefit multiscale simulations of mAb solutions by helping to incorporate flexibility into coarse grained models, ultimately aiding the design of more stable therapeutics.
Additionally, we have determined a method for free energy readout in the Fc domain, a target for modulating effector functions in vivo. Even though Fc is the “less flexible” IgG domain, its hinge and glycan chemistry dominate its conformational energetics. Removing the hinge or glycans lowers the free‑energy barrier for domain opening/closing, while retaining both features maximally stabilizes the dimer. The combined MD/PCA/umbrella‑sampling protocol offers a fast, practical, computational pipeline for predicting how Fc modifications will affect antibody stability and function.
- Quantify dynamics for mAbs. Protein flexibility underlies many biological functions—e.g., allosteric regulation, immune recognition, and antibody‑mediated therapeutics—but is notoriously hard to characterize, especially for large, multi‑domain proteins such as antibodies. Traditional experimental data (DEER, SAXS, etc.) are sparse, and the high‑dimensional conformational space of flexibly linked domains makes exhaustive modeling impractical. In conjunction with those experimental data, molecular dynamics simulations are used to generate structure ensembles of mAbs and their truncation mutants to “sample and select” conformations for a motional model. The resulting ensembles reveal multiple conformational states ranging from compact to highly extended geometries. The results suggest that intramolecular arrangements could influence intermolecular contacts, with implications for antibody function and aggregation control in biopharmaceuticals. Finally, the experimentally validated ensembles generated here provide valuable training data that could extend AI methods to predict dynamic, multi‑domain protein architectures.
Major Accomplishments
Publications/Presentations
Kleczynski, M., Bergonzo, C.*, Kearsley, A.J. Spatial and sequential topological analysis of molecular dynamics simulations of IgG1 Fc domains. J. Chem. Theory Computat. 21, 9, 4884-4897.
Szalai, V., Bergonzo, C.*, Lyon, R., Kelman, Z., Schmidt, T., Grishaev, A. Structure and dynamics of flexibly-linked, multi-domain proteins determined using spins, scattering, and simulations. 2025., Chem. Med. Chem. 20, 8, e202400917.
Hatch, H., Bergonzo, C., Blanco, M.A., Yuan, G., Grundinin, S., Lund, M., Curtis, J.E., Grishaev, A.G., Liu, Y., Shen, V.K. Anisotropic coarse-grain Monte Carlo simulations of lysozyme, lactoferrin, and NISTmAb by precomputing atomistic models. 2024, J. Chem. Phys. 161, 094113.
Bergonzo, C.*, Hoopes, J.T., Kelman, Z., Gallagher, D.T. Effects of glycans and hinge on dynamics in the IgG1 Fc. 2023 J. Biomol. Struct. Dyn. 1-9
Daniel R. Roe and Christina Bergonzo, PrepareForLeap: An Automated Tool for fast PDB-to-Parameter generation. 2022, J. Comp. Chem. 43, 13, 930-935.
Anderson, K. and Bergonzo, C., Scott, K., Karageorgos, I.L., Gallagher, E.S., Tayi, V.S., Butler, M., Hudgens, J.W. Hydrogen-deuterium exchange mass spectrometry and molecular dynamics simulations show glycoform dependent interactions between IgG1 and the FcgR1a receptor. 2021, J. Mol. Bio. Special Issue, Painting the mechanistic landscape of biomolecules. 434, 2, 167391.
Bergonzo, C. and Gallagher, D.T. Atomic model structure of intact NISTmAb. J. Res. NIST. 2021, 126.
“Classifying Biomolecular Structures through Unsupervised Learning Techniques” Spring ACS, CINF Section March 2024
“Applications of Open-Source Software to Biotherapeutic Platform Development” Fall ACS August 2023
“Using Molecular Dynamics to Advance Biotherapeutic Platform Development” Fall ACS August 2023
“Incorporating Dynamics to Advance Biotherapeutic Platform Development” LINXS Webinar June 2023
“Incorporating Dynamics to Advance Biotherapeutic Platform Development” Virginia Tech Biochemistry March 2023
“Building Computational Models to Advance Biotherapeutic Platform Development” NIST Biophysical and Biomedical Measurements Group Meeting February 2023