
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Atomic Reference Data for Electronic Structure Calculations, Reliability
Total energies
The standard deviation, σ, of the total energies for the calculations among the various codes in each of the four approximations is given as a function of atomic number Z in Fig. 1.
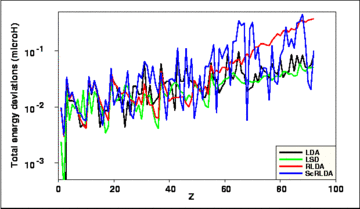
Fig. 1: Standard deviation of the total energies obtained by independent codes, as a function of atomic number Z, for the LDA, LSD, RLDA, and ScRLDA approximations as labelled.
The deviations exhibited in Fig. 1 are seen to increase somewhat with Z, but in no case do they exceed 0.5 microHartrees.
The actual differences between the results of the several codes are shown in the next four figures:
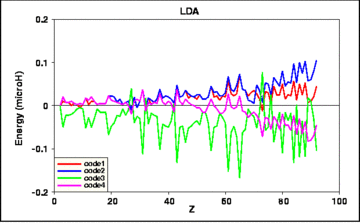
Fig. 2: Deviations of the results of 4 calculated LDA total energies from their average value, vs. atomic number Z.
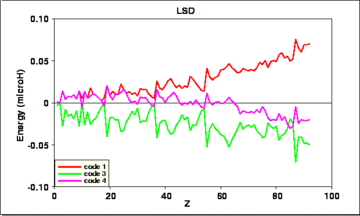
Fig. 3: Deviations of the results of 3 calculated LSD total energies from their average value, vs. atomic number Z.
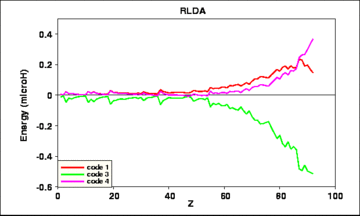
Fig. 4: Deviations of the results of 3 calculated RLDA total energies from their average value, vs. atomic number Z.
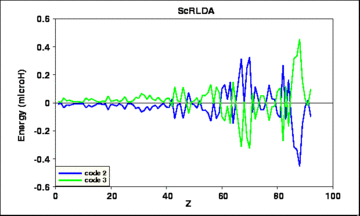
Fig. 5: Deviations of the results of 2 calculated ScRLDA total energies from their average, vs. atomic number Z.
Reliability: eigenvalues
Attainment of our basic criterion of convergence of total energies to microHartree accuracy has led to good agreement between independent calculations of the individual orbital energy eigenvalues. For the cases presented here, all individual orbital eigenvalues agreed to within 2 microHartrees.
- Reliability: total energies
- Reliability: eigenvalues
- Trends across the periodic table