
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Understanding Confined Fluids using Theory, Simulation, and Data-Driven Methods
Summary
Theory and simulation are used to advance our understanding of pure and multicomponent fluids confined in tight spaces, such as the pores of a zeolite, metal organic framework, shale, or sandstone. We develop and apply highly efficient theoretical and simulation methods to predict thermodynamic properties of confined fluids (e.g., adsorption isotherms, selectivities, Henry’s constants, heats of adsorption, etc.) and confined fluid phase behavior, in rigid and flexible frameworks. These tools are also employed to understand fundamentally the connection between dynamic properties and fluid structure. To this end, we explore the possibility of using equilibrium properties of confined fluids to predict their dynamic behavior over a broad range of pore loadings. In addition, we maintain the NIST/ARPA-e Database of Novel and Emerging Adsorbent Materials, a centralized resource for the scientific community to find and compare single- and multi-component adsorption isotherms reported in the literature.
Description
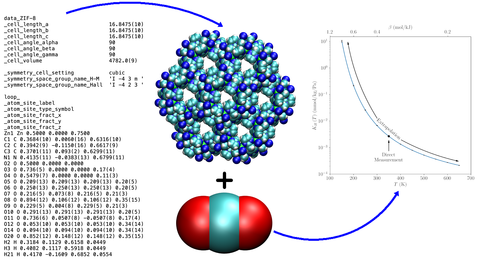
Thermodynamic tools allow estimation of the Henry Constant over a large temperature range from Crystal Structure
The most promising materials for capturing CO2 (or other greenhouse gases like CH4) from air or flue gas possess a large internal surface area per unit mass because they are composed of pores with diameters in the range of tens-to-hundreds of nanometers. Collectively referred to as mass separating agents (MSAs), these porous materials include adsorbents (e.g., metal-organic frameworks), polymer membranes, and combinations of the two (e.g., mixed matrix membranes). Once captured, terrestrial CO2 sequestration and mineralization processes involve fluid in contact with porous sedimentary rock, such as shale, for storage or subsequent chemical transformation. These rock formations are composed of complex and chemically heterogeneous pore networks and contain other chemical species like water and hydrocarbons. Clearly, understanding the behavior of pure CO2 in confined environments, such as those afforded by these different materials is critical. Furthermore, when a fluid is confined to spaces comparable to molecule dimensions, its properties can be dramatically altered from those of the bulk due to the increased surface area exposed to the confining environment (e.g., pore wall); as a result, existing measurements, data, and equations of state for bulk fluids are not applicable in these situations. In addition, CO2 is rarely encountered as a pure fluid in practice. Thus, separation scientists and engineers seek properties of confined CO2 mixtures when identifying candidate MSAs and developing processes for gas capture and sequestration. Unfortunately, given the diverse range of possible feed conditions (e.g., composition, temperature, and density) and material types (e.g., adsorbents, membranes, and porous rocks), comprehensive experimental characterization of this parameter space is a daunting task without a coordinated effort involving multiple entities; such an effort does not exist. Thus, there is a large knowledge gap in the development of carbon and greenhouse gas capture and sequestration processes.
Our research efforts employ theory and simulation to study and predict the behavior of pure and multicomponent fluids in confinement. The scope of our work extends to greenhouse gas capture and sequestration processes, as well as to the broader topic of MSA-based separations which require less energy than conventional chemical distillation. In addition to studying confined pure fluids, such as CH4, CO2, and H2O, we develop advanced simulation methods to predict binary adsorption isotherms under a variety of feed conditions, which in turn provide selectivities and heats of adsorption, key pieces of information separation scientists and engineers need to identify candidate MSAs. The fluid phase behavior of confined mixtures, which is difficult to determine experimentally, is also a topic we address with simulation, since this is an important issue in sequestration processes where residual water and hydrocarbons can be encountered, potentially leading to transport issues and pore blockage. Dynamic properties such as the confined self-diffusivity are important but require substantially more computational and experimental effort. Here, we have developed scaling relations that allow one to predict the self-diffusivity as a function pore loading. Extending this to multicomponent fluids is the next step in this area of work.
Ultimately, we seek to use theory and simulations to help fill the substantial knowledge gap regarding the properties of confined fluid mixtures. In addition, this data can be fed into process simulation software for exploratory design and can serve as the basis for advancing thermodynamic theories of confined fluids (i.e., beyond approximations such as Ideal Adsorbed Solution Theory) or for training AI models. Fundamentally and crucially, simulations provide insight into the molecular-level processes and mechanisms (e.g., gating) that determine selectivity, capacity, and transport in porous materials. Knowledge of this type can ultimately be used to rationally design MSAs with desired pore geometries and pore chemistry.
Other Related Projects
FEASST: Free Energy and Advanced Sampling Simulation Toolkit
NIST Data Resources for Adsorption Science and Technology
Associated Publications
1. Siderius, D.W., Hatch, H. W., and Shen, V. K., "Temperature Extrapolation of Henry's Law Constants and the Isosteric Heat of Adsorption," The Journal of Physical Chemistry B, 126, 7999 (2022).
2. Siderius, D. W., "Digitization of Adsorption Isotherms from" The Thermodynamics and Hysteresis of Adsorption"," Journal of Research of the National Institute of Standards and Technology, 126, 1-7 (2021).
3. Ongari, D., Talirz, L., Jablonka, K. M., Siderius, D. W., and Smit, B., "Data-Driven Matching of Experimental Crystal Structures and Gas Adsorption Isotherms of Metal–Organic Frameworks," Journal of Chemical and Engineering Data, 67, 1743, (2022).
4. Wong-Ng, W., McCandless, G. T., Culp, J. T., Lawson, M., Chen, Y. S., Siderius, D. W., Chen, Y. P., and Li, L., "Crystal structure, sorption properties, and electronic structure of flexible MOF, (Ni-4, 4’ azopyridine)[Ni(CN)4]," Solid State Sciences, 118, 106646 (2021).
5. Rzepa, C., Siderius, D. W., Hatch, H. W., Shen, V. K., Rangarajan, S., and Mittal, J., "Computational investigation of correlations in adsorbate entropy for pure-silica zeolite adsorbents," The Journal of Physical Chemistry C, 124, 16350-16361 (2020).
6. Cook, L. P., Brewer, G. A., Siderius, D., and Wong-Ng, W., "Topology of voids and channels in selected porphyrinic compounds," Powder Diffraction, 34, 302-310 (2019).
7. Krekelberg, W. P., Mahynski, N. A., and Shen, V. K., "On the virial coefficients of confined fluids: Analytic expressions for the thermodynamic properties of hard particles with attractions in slit and cylindrical pores to second order," Journal of Chemical Physics, 150, (2019).
8. Sturluson, A., Huynh, M. T., Kaija, A. R., Laird, C., Yoon, S., Hou, F., Feng, Z., Wilmer, C. E., Col+¦n, Y. J., and Chung, Y. G., "The role of molecular modeling and simulation in the discovery and deployment of metal-organic frameworks for gas storage and separation," Molecular Simulation, 45, 1082-1121 (2019).
9. Shen, V. K., Siderius, D. W., and Mahynski, N. A., "Molecular simulation of capillary phase transitions in flexible porous materials," The Journal of Chemical Physics, 148, 124115 (2018).
10. Wong-Ng, W., Nguyen, H. G., Espinal, L., Siderius, D. W., and Kaduk, J. A., "Powder X-ray structural studies and reference diffraction patterns for three forms of porous aluminum terephthalate, MIL-53 (A1)," Powder Diffraction, 34, 216-226 (2019).
11. Krekelberg, W. P., Siderius, D. W., Shen, V. K., Truskett, T. M., and Errington, J. R., "Connection between thermodynamics and dynamics of simple fluids in pores: Impact of fluid fluid interaction range and fluid solid interaction strength," The Journal of Physical Chemistry C, 121, 16316-16327 (2017).
12. Krekelberg, W. P., Siderius, D. W., Shen, V. K., Truskett, T. M., and Errington, J. R., "Position-dependent dynamics explain pore-averaged diffusion in strongly attractive adsorptive systems," Langmuir, 33, 13955-13963 (2017).
13. Siderius, D. W., Mahynski, N. A., and Shen, V. K., "` between pore-size distribution and flexibility of adsorbent materials: statistical mechanics and future material characterization techniques," Adsorption, 23, 593-602 (2017).
14. Siderius, D. W., Krekelberg, W. P., Chiang, W. S., Shen, V. K., and Liu, Y., "Quasi-Two-Dimensional Phase Transition of Methane Adsorbed in Cylindrical Silica Mesopores," Langmuir, 33, 14252-14262 (2017).
15. Gor, G. Y., Siderius, D. W., Shen, V. K., and Bernstein, N., "Modulus-pressure equation for confined fluids," Journal of Chemical Physics, 145, (2016).
16. Mahynski, N. A. and Shen, V. K., "Multicomponent adsorption in mesoporous flexible materials with flat-histogram Monte Carlo methods," The Journal of chemical physics, 145, (2016).
17. Ross, R. B., Aeschliman, D. B., Ahmad, R., Brennan, J. K., Brostrom, M. L., Frankel, K. A., Moore, J. D., Moore, J. D., Mountain, R. D., and Poirier, D. M., "Adsorption, X-ray diffraction, photoelectron, and atomic emission spectroscopy benchmark studies for the eighth industrial fluid properties simulation challenge," Adsorption Science & Technology, 34, 13-41 (2016).
18. Siderius, D., Shen, V. K., Mountain, R. D., Schultz, N. E., Ahmad, R., Brennan, J. K., Frankel, K. A., Moore, J. D., Ross, R. B., and Thommes, M., "The Eighth Industrial Fluid Properties Simulation Challenge," (2016).
19. Gor, G. Y., Siderius, D. W., Rasmussen, C. J., Krekelberg, W. P., Shen, V. K., and Bernstein, N., "Relation between pore size and the compressibility of a confined fluid," The Journal of chemical physics, 143, (2015).
20. Wong-Ng, W., Kaduk, J. A., Siderius, D. L., Allen, A. L., Espinal, L., Boyerinas, B. M., Levin, I., Suchomel, M. R., Ilavsky, J., and Li, L., "Reference diffraction patterns, microstructure, and pore-size distribution for the copper (II) benzene-1, 3, 5-tricarboxylate metal organic framework (Cu-BTC) compounds," Powder Diffraction, 30, 2-13 (2015).
21. Ross, R. B., Brennan, J. K., Frankel, K. A., Moore, J. D., Moore, J. D., Mountain, R. D., Ahmad, R., Thommes, M., Shen, V. K., and Schultz, N. E., "Perfluorohexane adsorption in BCR-704 Faujasite zeolite benchmark studies for the seventh industrial fluid properties simulation challenge," Fluid Phase Equilibria, 366 , 141-145 (2014).
22. Ross, R. B., Ahmad, R., Brennan, J. K., Frankel, K. A., Moore, J. D., Moore, J. D., Mountain, R. D., Shen, V. K., Schultz, N. E., and Siderius, D. W., "The seventh industrial fluid properties simulation challenge," Fluid Phase Equilibria, 366, 136-140 (2014).
23. Shen, V. K. and Siderius, D. W., "Elucidating the effects of adsorbent flexibility on fluid adsorption using simple models and flat-histogram sampling methods," The Journal of chemical physics, 140, (2014).
24. Krekelberg, W. P., Siderius, D. W., Shen, V. K., Truskett, T. M., and Errington, J. R., "Connection between thermodynamics and dynamics of simple fluids in highly attractive pores," Langmuir, 29, 14527-14535 (2013).
25. Mountain, R. D., "Molecular dynamics simulation of water acetonitrile mixtures in a silica slit," The Journal of Physical Chemistry C, 117, 3923-3929 (2013).
26. Siderius, D. W. and Shen, V. K., "Use of the grand canonical transition-matrix Monte Carlo method to model gas adsorption in porous materials," The Journal of Physical Chemistry C, 117, 5861-5872 (2013).
27. Krekelberg, W. P., Shen, V. K., Errington, J. R., and Truskett, T. M., "Impact of surface roughness on diffusion of confined fluids," Journal of Chemical Physics, 135, (2011).
28. Goel, G., Krekelberg, W. P., Pond, M. J., Mittal, J., Shen, V. K., Errington, J. R., and Truskett, T. M., "Available states and available space: Static properties that predict self-diffusivity of confined fluids," Journal of Statistical Mechanics: Theory and Experiment, 2009, 04006 (2009).
29. Mittal, J., Shen, V. K., Errington, J. R., and Truskett, T. M., "Confinement, entropy, and single-particle dynamics of equilibrium hard-sphere mixtures," The Journal of chemical physics, 127, (2007).