
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
HDX-MS for Biopharmaceutical Analysis
Summary
This project applies hydrogen/deuterium exchange mass spectrometry (HDX-MS) for determining the structural dynamics of biotherapeutics, glycoproteins, membrane proteins, and other proteins and their interactions. Topics of particular interest include protein-ligand interactions, structure-function relationships between glycan structure and glycoprotein folding, comparability measurements among innovator drugs and biosimilars, and interlaboratory comparisons. This project aims to improve measurements through method, instrumentation, and software development.
Description
This project is designed to improve, test, and validate hydrogen/deuterium exchange mass spectrometry (HDX-MS) metrology for the determination of dynamical properties of therapeutic proteins and glycoproteins. The research project uses HDX-MS to measure the D-for-H exchange rates of the amide groups along the backbone of a protein in D2O solution. These rates indicate the protection factors of the amide groups. The protection factors are characteristic of higher order structural features of proteins that are stabilized through hydrogen bonding, disulfide bonds, electrostatic interactions, and hydrophobic forces. Since protection factors can change as the protein binds with ligands or undergoes folding, HDX-MS can provide sensitive diagnostic data evidencing structural differences. HDX-MS is a rapidly evolving metrology. Numerous improvements in automation, instrumentation, resolution, accuracy, and software are being introduced.
Objectives
- Explore structure-function relationships between glycan structure and protein folding energies
- Develop HDX-MS for assessments of dynamical comparability among innovator and candidate biosimilar drugs
- Measure the reproducibility of proteolytic fragmentation HDX-MS by conducting interlaboratory comparisons on a Fab protein
- Improve HDX-MS technology for measurements of transmembrane protein drug targets
- Measure interactions of biological drugs with surfaces and aggregates
- Enhance resolution of the HDX-MS method to the single amide level
- Adopt internal H/D exchange reporters and materials to become NIST standard reference materials for improvement of reproducibility and harmonization of HDX-MS
Postdoctoral Research Opportunities
National Research Council Postdoctoral Research Associate Program at NIST: Rolling application deadlines are Feb 1 and Aug 1. US citizens only. $82,764 (2025) annual salary plus benefits, $3,000 travel allotment, and research funding . Contact kyle.anderson [at] nist.gov (Kyle Anderson) before applying. View Job Posting.
Positions for non-US citizens: Contact kyle.anderson [at] nist.gov (Kyle Anderson) directly.
Major Accomplishments
HDX-MS Interlaboratory Comparison Project
An interlaboratory study involving 37 co-authors in 15 unharmonized laboratories determined the reproducibility of continuous-labeling, bottom-up HDX-MS measurements by measuring the deuterium uptake of Fab of NISTmAb (PDB: 5K8A) for immersions in D2O for tHDX = (30 to 14400) s and THDX = (25, 21, 3.6) oC. Each laboratory was sent a standardized kit containing Fab protein and the reagents necessary to conduct experiments.
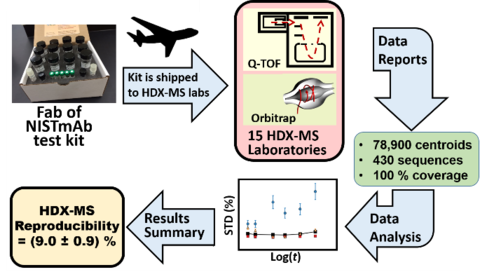
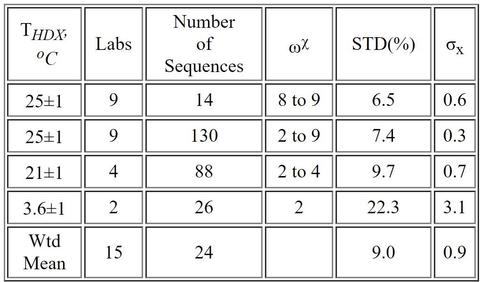
Laboratories reported ≈89,800 centroid measurements for 430 proteolytic peptide sequences of the Fab fragment (≈78,900 centroids) and ≈10,900 centroid measurements for 77 peptide sequences of the Fc fragment. Nearly half of peptide sequences were unique to the reporting laboratory, and only two sequences were reported by all laboratories. The majority of the laboratories (87 %) exhibited centroid mass laboratory repeatability precisions of ≤ (0.15 ± 0.01) Da. All laboratories achieved ≤ 0.4 Da. For immersions of protein at THDX = (3.6 to 25) °C and for D2O exchange times of tHDX = 30 s to 4 h, the reproducibility of back-exchange corrected deuterium uptake measurements for the 15 laboratories was (9.0 ± 0.9) %. A 9 laboratory cohort that immersed samples at THDX = 25 °C exhibited reproducibility of (6.5 ± 0.6) % for back-exchange corrected deuterium uptake measurements.
Automated Removal of Phospholipids from Membrane Proteins
NIST researchers developed an automated approach to study the structural dynamics of membrane proteins, the most common targets for pharmaceuticals. The method involves isolation of proteins from their phospholipid bilayer using a scheme that is compatible with robotic handling. The key development was the introduction of ZrO2 microbeads, which strongly coordinate with the membrane phospholipids, and then removal of microbead-coordinated phospholipids by inline nanofilters. The protein is thus freed from phospholipids, which interfere with analysis by LC-MS.
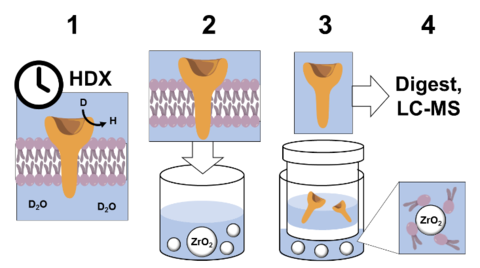
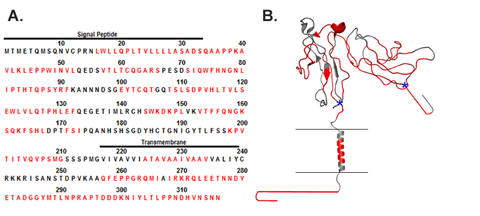
Previous HDX-MS publications on membrane protein dynamics removed phospholipids manually, requiring arduous, hours-long laboratory work. The new approach was demonstrated on the transmembrane protein FcγRIIa, and showed information about the regions outside of, across, and inside of its liposomal membrane, enabling measurement of structural dynamics of the whole protein. This approach is the first to enable fully automated HDX-MS on full-length transmembrane proteins in lipid bilayers.
HDX-MS Subzero Chromatography System for Reduced Back-exchange and Rapid Removal of Sample Carryover
New applications for HDX-MS, particularly those involving quality control and biosimilarity evaluations, will demand deuterium uptake measurements of improved precision. NIST researchers developed an analysis system for HDX-MS that conducts chromatography at ‑30 °C. Extending gradients to 40 minutes at -30 °C exhibited ≈16 % more deuterium recovery than for a conventional 8 minute gradient at 0 °C, and a subset of peptides showed ≈26 % more deuterium. Chromatographic resolution is improved, allowing HDX-MS measurements on a greater number of peptides. Total measurement uncertainty is greatly reduced. Reproducibility is enhanced by regulating chamber temperatures to ±0.05 °C. Isotopic bias and sample carryover is effectively eliminated with efficient cleaning cycles that include backflushing of all columns and an added wash pump to provide FOS-choline-12 (n-dodecylphosphocholine) in water, which rapidly removed carryover from NISTmAb and reduced necessary blanks from 4 to 1. The analysis system stores two enzyme/protease columns that are perpetually cleaned and always ready for use. Methods were developed for both reversed phase and HILIC subzero temperature chromatography with different mobile phase options to maximize platform flexibility and facilitate adoption.

