
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Taking Measure
Just a Standard Blog

In December 2013, NIST group members wrote their predictions on the value of Planck’s constant they would measure on their NIST-4 Kibble balance. Shisong Li, a guest researcher from Tsinghua University in China, came closest. His prediction differed by only about 5 parts per billion from the measured result.
So, a bunch of metrologists walk into a bar.
In late December 2013, a group of NIST researchers met at the local watering hole across the street from NIST’s Gaithersburg, Maryland, campus. NIST researchers meeting up after work during the holidays is a common occurrence, but this particular group had more on their minds than just hoisting a few pints. As it turns out, they had been working on hoisting something else.
The group had good reason to celebrate that evening. They had just achieved a major milestone in their work and were looking forward to the next phase of their research.
“I remember being at the long table on the second floor of the bar passing around this napkin,” says mechanical engineer Leon Chao.
That napkin was the beginning of a bar bet concerning a fundamental constant of the universe—a bet that would take four more years to settle.
Written on the napkin were several long strings of numbers set equal to the letter h, a symbol known well to Chao and his fellow researchers for denoting Planck’s constant. The many digits were central to the work they were celebrating: NIST’s contribution to a worldwide scientific effort to redefine exactly what a kilogram is.

Big K and little h
The International Prototype Kilogram (IPK) is a shiny cylinder of platinum and iridium. Cast more than 120 years ago, it lives a closely guarded life inside a triple-locked underground vault outside Paris. Affectionately known as Le Grand K, or Big K, this metal cylinder is the kilogram. It’s the king when it comes to measuring mass. All measurements of weight and mass in practically every country on Earth can be traced back, through national metrology institutes like NIST, to Big K.
But it’s not a perfect system. Just imagine if Big K was dropped. Or stolen. Or breathed on.
A long-term endeavor of many measurement scientists around the world is to improve the system by replacing Big K. Not with another object, but with a NIST equation based on unchanging constants of the universe. Much like the redefinition of other basic units of measurement, such as the meter and the second, the aim is to define the standard of mass in terms of physical constants instead of a fragile, filchable object.
The linchpin for achieving this ambitious goal is determining a hyper accurate value for h, Planck’s constant, a number identified by physicist Max Planck in 1900, back when Big K was only 11 years old. Through the relationship of E=hν, Planck’s constant (h) links a single photon’s teeny tiny amount of energy (E) to its frequency (ν). By combining it with Einstein’s discovery of the equivalence of mass and energy—represented by the more famous E=mc2—a kilogram can be precisely defined in terms of energy, rather than by a vault-dwelling metal cylinder in France.
But to truly dethrone Big K, researchers would have to determine the value of Planck’s constant to an almost unthinkable level of accuracy, equivalent to measuring the distance between New York City and Washington, D.C., down to less than 1.27 centimeters (0.5 inch). This level of accuracy would not be even remotely possible until more than a century after Planck’s discovery.
Meanwhile, back at the bar
The NIST researchers enjoying happy hour in December 2013 were celebrating their recent achievement—a newly submitted and highly accurate value of Planck’s constant, achieved using a complex instrument called a watt balance, which is now called a Kibble balance, in honor of the device’s inventor, Bryan Kibble. NIST’s watt balance program goes back many years and several generations of instruments.

The basic function of a watt balance is similar to a traditional balance scale with two pans, but with a critical difference. Where a balance scale compares two masses, a watt balance compares the weight of a mass with an electromagnetic force. In 2013, NIST was on its third-generation watt balance, NIST-3, a liquid-helium-guzzling beast of a machine that researchers kept confined to a multistory copper-lined cage.
Although NIST-3 was a high-precision instrument, the researchers at the bar knew it was not accurate enough to redefine the kilogram. To attain that level of accuracy, they would need to build a new watt balance: NIST-4. Their hope was that NIST-4 would provide the most accurate measurement yet of Planck’s constant and, when combined with research at other national metrology institutes, finally provide a new definition of the kilogram and give Big K a well-earned retirement.
So, how to commemorate this important, nay, historic, occasion? With a friendly bar bet, of course.
Anybody got a pen?
“Everyone was very excited,” recounts Frank Seifert, a NIST guest researcher. “We had just submitted our data from NIST-3 and we were looking forward to building NIST-4.”
Physicist Stephan Schlamminger was there as well. “That night we went out to the bar for happy hour,” he remembers. “Sitting around the table, it came up spontaneously. Dave (Newell, another NIST physicist) had a pen and started writing.

Each of the researchers took a crack at guessing the final value of Planck’s constant that NIST would eventually determine and submit. They were looking ahead several years into the future, to the time when Planck’s constant would be defined so accurately that it could be used, along with other physical constants, to truly redefine the kilogram.
Of course, it was also happy hour.
“I thought, ‘I’m going to win it,’” says physicist Darine Haddad. “I was sitting there drinking my beer, doing the calculation ... I think I missed a nine halfway through my beer.”
The result of all this was an undetermined number of pints consumed and a napkin with 10 individual guesses for the value of h, one from each person who contributed to the work.
Now, if you’d like to examine this very mathy napkin, you’re out of luck. The researchers sealed it inside a plastic bottle and buried it that Christmas Eve in a suitable location: under about 1.82 meters (6 feet) of sand in a cavity within the massive concrete foundation of NIST-4.
And there it will remain, a permanent part of the project’s foundation both literally and figuratively, perhaps to be discovered by future metrologists.

A piece of cake
Fast-forward four years ahead to the summer of 2017. The watt balance team, using NIST-4, has now submitted yet another value for Planck’s constant, this one more accurate than any previous value determined by NIST.
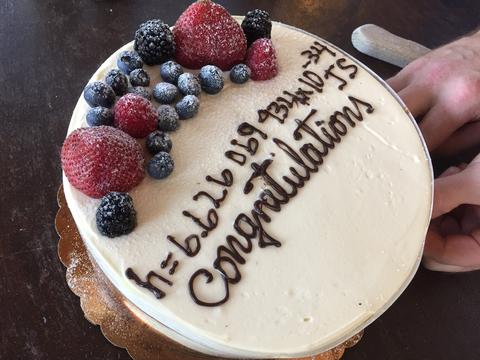
So, who came the closest and won the bet? That honor goes to Shisong Li, in 2013 a guest researcher at NIST from China’s National Institute of Metrology. Li now works in France, quite near to Big K, at the International Bureau of Weights and Measures.
NIST’s newly submitted value for Planck’s constant is now being compared with values submitted from the national measurement labs of other countries. Their combined work will be used to permanently fix the value of Planck’s constant, defining it with a level of precision that likely would’ve surprised Planck himself.
And, as you might imagine, the researchers made another well-earned pilgrimage to the bar to celebrate. This time, however, they had a fancy cake to commemorate the occasion. Among the decorations on top of the cake is NIST’s final number for h, written in icing: 6.626069934 x 10−34 kg∙m2/s.
But it’s much more impressive when written like this:
0.0000000000000000000000000000000006626069934 kg∙m2/s
And it was an Italian rum cake, by the way.
About the author

Related Posts



Comments
Hi Stephen,
Stephan Schlamminger, one of the physicists mentioned in the post, published a graph showing the latest values side-by-side on his personal website. Regarding the Scientific American article, I imagine that there will be more to come in the next couple years on this, but in the meantime, you can read this one from earlier this year.
Hope this helps!
Mark Esser
Editor, Taking Measure blog
Поздравляю!
On behalf of the researchers, thank you. до свидания!
Great article, but I thought the symbol for the Planck constant was h-bar, not h as used in the article, napkin and cake.
I’m not at all qualified to answer this question, but a NIST physicist (who is) told me the following: “The symbol h denotes the Planck constant. The symbol h-bar is an abbreviation for h divided by 2 pi.”
A photon obeys the principle of least action and seeks a path around mass with as big steps (oscillations of low frequency) as possible and a minimum of these. Observed is a slightly parabolic route which means that closer to the mass time runs faster or oscillations have higher frequency. This is explained by Vasily Yanchilin in his book The Quantum Theory of Gravitation (2003). Fast processes at the Beginning correspond with it but not existence of so called black holes. However atomic clocks in satelites tick faster than on Earth if measurements are correct.
Yanchilin thinks that light may generate dark mass since radiation is everywhere. To this can be added that dispersed radiation does not react anymore with anything but its energy cannot get lost. If it turns into dark mass particles these may gradually escape into space, but temporarily a sphere of dark mass might be around Earth, raising local potential and make those atomic clocks in space speed up. How should be measured the amount of radiation that got old and dispersed, does not react anymore with common particles? Perhaps a computer model is possible. The observation of the photon's route is not in agreement with the general theory of relativity and thus the Planck based on that theory of a century ago cannot be trusted to be constant. According Yanchilin its value increases with the expansion of the universe. If so it is a questionable base for the kilogram. It should be noted that c was bigger int he past if the Planck then was smaller and therefore the standard supernova Ia has to be corrected. Then accellerated expansion of the universe disappears and negative energy becomes superfluous. NIST may give a lecture on the principle of least action and its consequences.
Boldly taking lawn mower safety where no man has gone before!
We boldly measure what no one has measured before.
How close to the other Kibble balance measurements are you? When will you have a Scientific American article?
Congratulations!!