
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Nathan A. Mahynski (Fed)
Chemical Engineer

As technology grows ever more complex in the modern era, so-called “soft matter” systems play an increasingly important role at the heart of many technologies and across a plethora of scientific disciplines. My background is in computational thermodynamics and my research at NIST focuses on three principal areas, each broadly aimed at addressing certain challenges which arise from attempts at understanding, modeling, and predicting the behavior of soft matter systems:
- Developing new computational algorithms to simulate fluids and understand the self-assembly of molecular and macromolecular systems, such as colloids and polymers.
- Using computational crystallography and related methods to predict the solid / crystalline behavior of these systems.
- Applying machine learning / artificial intelligence methods to predict, authenticate, and understand patterns in food, food products, and related biological systems.
A more detailed description of selected topics can be found below. For instance, using computer simulations and machine learning to understand the directed assembly of DNA-grafted colloids, multicomponent fluid adsorption in flexible materials, and developing efficient "extrapolation" methodologies for simulations and data analysis. For research openings and opportunities, please email me at nathan.mahynski [at] nist.gov (Subject: Research%20Inquiry) (nathan[dot]mahynski[at]nist[dot]gov). Currently available positions include:
-
Guest researcher: the project will focus on the use of machine learning and inverse design to assemble multicomponent colloidal crystals with complexity ranging from single component systems to those with addressable complexity. The successful applicant should be well-versed in the Python programming language. nathan.mahynski [at] nist.gov (Subject: Research%20Inquiry) (Contact me) for more details.

-
NRC Postdoctoral Fellowship: Research Opportunity #50.64.61.C0449 in computational self-assembly - see the here for more details. Note that applicants must be have US citizenship.
The referenced media source is missing and needs to be re-embedded. -
Summer Undergraduate Research Fellowship (SURF): these are excellent research opportunities for undergraduate students across many different disciplines to obtain hands-on experience doing top-tier research. Many students have successfully published peer-reviewed papers based on their summer work here. Interested students should nathan.mahynski [at] nist.gov (Subject: Research%20Inquiry) (contact me) for current offerings. See NIST's SURF website for more details about the program.
Programs/Projects
- Classification Methods for the NIST Biorepository
- Machine Learning Fluid Equations of State
- Machine Learning to Predict Multicomponent Colloidal Crystals
- Machine Learning to Predict Food Provenance
- FEASST: The Free Energy and Advanced Sampling Simulation Toolkit
- PACCS: Python Analysis of Colloidal Crystal Structures
Recent Highlights



Awards
- 2020 MML Accolade
- NIST Sigma Xi PPP Outstanding Poster in Materials (2016), and Computer Modeling and Simulation & Physics (2017)
- Univ. of Washington Distinguished Young Scholar in Chemical Engineering, 2016
- Christopher J. Wormald Prize in Thermodynamics, 2015
- NRC Research Associateship Program Fellowship, 2015
- Gordon Wu Prize for Excellence at Princeton University, 2014
Memberships
- American Institute of Chemical Engineers (AIChE), 2007-Present
- American Physical Society (APS), 2019-Present
- Tau Beta Pi, 2010-Present
- Sigma Xi, 2017-present
Research
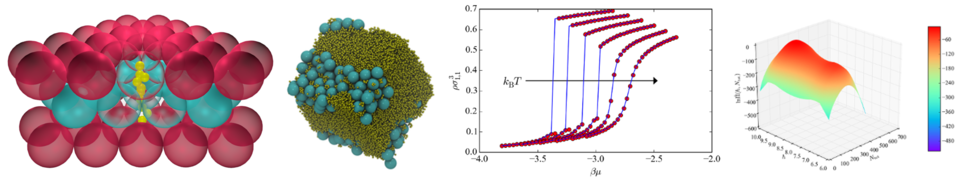
Colloidal Self-Assembly
Globally ordered colloidal crystal lattices have broad utility in a wide range of optical and catalytic devices, for example, as photonic band gap materials. However, the self-assembly of stereospecific structures is often confounded by polymorphism. Small free-energy differences often characterize ensembles of different structures, making it difficult to produce a single morphology at will. Current techniques to handle this problem adopt one of two approaches: that of the "top-down" or "bottom-up" methodology, whereby structures are engineered starting from the largest or smallest relevant length scales, respectively. However, recently our work has focused on developing a new methodology for controlling colloidal crystal assembly and polymorphism using external structure-directing agents and symmetry-based computational algorithms. To this end, special attention has been paid to designing algorithms for predicting crystal polymorphism and the relative stability of multicomponent colloidal mixtures. Click the image below, or the links at the bottom of the page to see some recent publications.
Computational Methods for Data-driven Thermodynamics
With the advent of the fourth paradigm of science ("big data"), data-intensive, if not data-driven, approaches are proving to be novel, effective tools for scientific inquiry. In contrast to the previous paradigm ("computational"), for many areas of research, data-driven approaches rely less on small amounts of very accurate data in favor of larger amounts of "reasonably accurate" data to learn things about the natural world. In the field of chemical thermodynamics, a large body of work has emerged over the past half century describing highly accurate, but often computationally intensive, simulation tools for investigating the thermodynamic behavior of fluids. Central to this is the focus on increasingly complex schemes to provide more accurate calculations regardless of the expense. Such solutions do not scale well when trying to investigate a wide range of systems or cases. My work focuses on revisiting early ideas from the field of statistical mechanics, and developing some new ones, to create fast, approximate methods that permit high-throughput, data-driven investigation of the thermodynamic properties of fluids, as well as predicting complex, alchemical transformations.
In molecular simulation, "alchemical" transformations refer to the mutation of one aspect of a molecule or atom from one state into another. For example, transmuting a monomer bead on a block copolymer from type "A" to "B", or the residue on a protein from one amino acid to another. Knowledge of the free energy change, or other thermodynamic properties, associated with these mutations can be key to assessing the behavior of many physical systems and notably in the optimization of ones with many interchangeable "parts" or variables, such a protein or other complex chemical. If a well-defined set of coordinates, or path, is known which describes the system changing from one state to another, there exist numerous computational methods for estimating the free energy change associated with this alchemical transformation. However, the computational cost associated with many of these methods often presents a barrier to their widespread application in, e.g., optimization of pharmaceutical molecules. Furthermore, it is generally prohibitively expensive to compute enough information about enough permutations of different variables to enable data-driven approaches, such as machine learning, to be brought to bear. To remedy this, ongoing work with various collaborators focuses on developing fast, approximate alternatives that enable these calculations to be performed quickly and efficiently enough to enable data-centric approaches.
Alternative Separation Technologies
Chemical separations in the manufacturing industry account for as much as 15% of global energy use, and as much as half of the industrial energy used in the U.S. While porous mass separating agents (MSAs) appear to be one plausible lower energy alternative, the lack of certain fundamental relationships between the behavior of fluid mixtures confined in porous materials and the selectivity of MSA-based processes presents a significant barrier to their widespread industrial application. Reducing the energy required to achieve separation of industrially-relevant fluids is essential to ensuring the sustainability of the chemical manufacturing sector. In collaboration with the American Chemical Society's Green Chemistry Institute Chemical Manufacturers Roundtable, my work focuses on developing theoretical and computational tools which will help accelerate the industrial application of less energy-intensive chemical separation alternatives to distillation. Most of the efforts by myself and coworkers is focused on understanding fundamental aspects of outstanding questions in adsorption-based MSA processes in order to enable the rational design of such processes in the future.