
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
We use machine learning and other computational techniques to predict the self-assembly characteristics of multicomponent colloidal materials. This is a particularly challenging prediction problem due to the breadth of possible interactions that colloidal systems can achieve, the combinatorial explosion of possible crystalline forms with unit cell size, and the large number of possible constituents. By leveraging a combination of mathematical symmetry and computational techniques including ML, we aim to provide a predictive framework for experimentally realizable synthesis routes to targeted colloidal structures.
Description
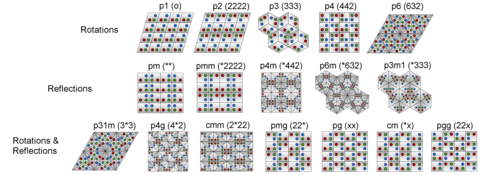
Classification of different types of symmetry relevant to machine learning computational techniques.
There is a direct link between a material’s macroscopic properties and its microscopic structure, which makes rational bottom-up self-assembly a powerful tool for engineering properties of materials. In general, colloids are facile material building blocks whose shape, charge, and surface functionalization can be tuned to control their assembly. However, the breadth of this design space usually requires computational approaches to predict the outcome of an assembly process. The technique of choice is generally ‘‘inverse design’’ in which a chosen target structure is sought by systematically reverse engineering the interactions (chemistry) of the colloids. This is typically achieved through an optimization procedure, such as one based on relative entropy.
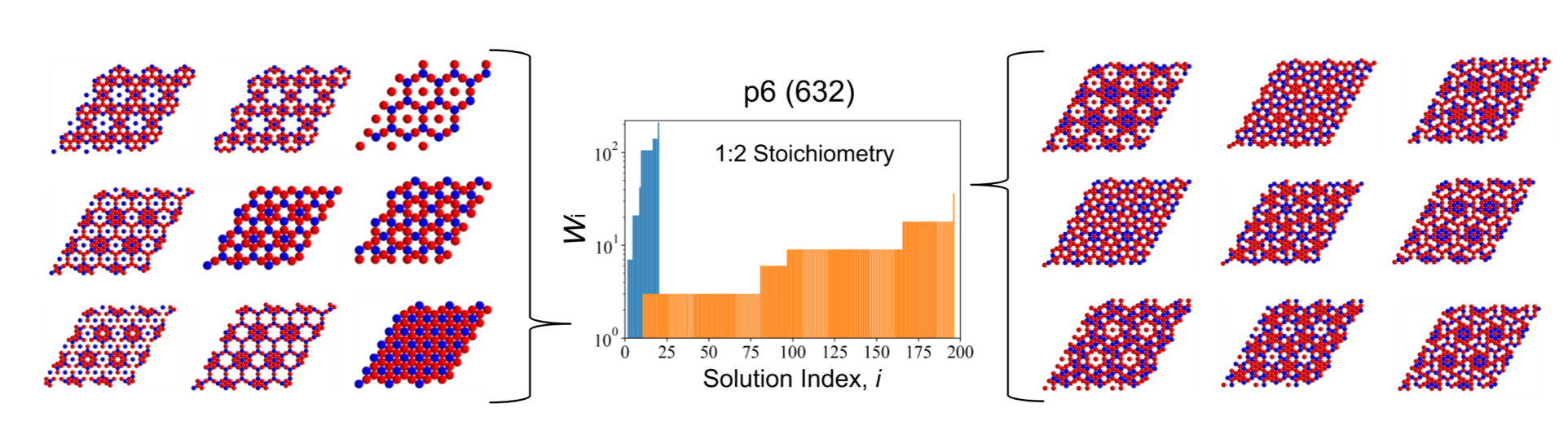
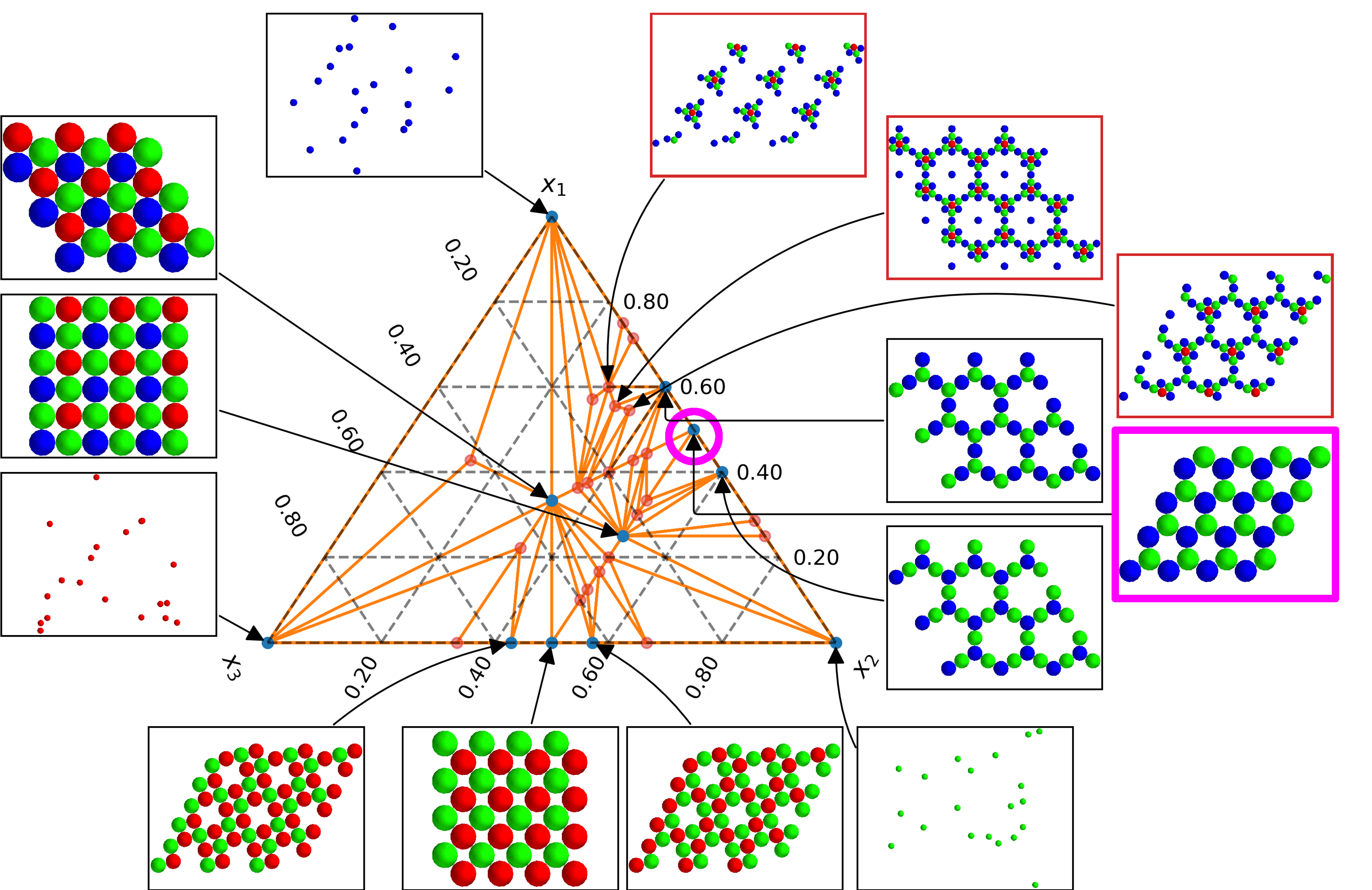
We have recently developed an approach for systematically enumerating crystalline ground states of two-dimensional multicomponent colloidal materials using symmetry. [1,2] This allows us to generate phase diagrams for multicomponent mixtures which govern their equilibrium self-assembly, by searching over many, if not nearly all, relevant competing crystalline lattices. Importantly, this scheme enables inverse design while explicitly accounting for the possibility of phase separation by wrapping this search in a machine learning algorithm. [3] Conventional inverse design methods typically optimize pairwise interactions in a canonical (NVT) system to yield a Hamiltonian that will best assemble into a desired structure at that same composition. While powerful, this can struggle to handle cases when phase separation occurs; moreover, in single component systems the apparent complexity of a chosen target’s structure tends to be reflected in the resulting pairwise interactions, making these optimized systems difficult or impossible to realize experimentally. One way to produce more realizable systems is to rely on mixtures of components with simpler interactions, rather than on a single component with a more complicated interaction.
This project will use theory, simulation, and machine learning to understand how chemical functionalization of soft matter systems (e.g., colloids, polymers) affects their self-assembly into different crystals or other morphologies, and how this is related to their equilibrium phase behavior. This will explore the application of powerful, modern ML approaches to achieve inverse design more accurately, in less time, and in an experimentally viable fashion. Previous work will be extended to connect it with experimentally viable design routes enabling the rational design and synthesis of colloidal crystals, monolayers, and functional materials. These symmetry considerations will especially be used to derive functionality for self-assembling nanoscale frameworks.
Major Accomplishments
PACCS code for a colloidal crystal structure analysis, generation, optimization, and visualization library written in Python is freely available on gihub.
References
- “Using symmetry to elucidate the importance of stoichiometry in colloidal crystal assembly,” N. A. Mahynski, E. Pretti, V. K. Shen, J. Mittal, Nature Commun., 10 2028 (2019).
- “Symmetry-based crystal structure enumeration in two dimensions,” E. Pretti, V. K,'" E. Pretti, V. K. Shen, J. Mittal, N. A. Mahynski, J. Phys. Chem. A 124, 3276-3285 (2020).
- “Grand canonical inverse design of multicomponent colloidal crystals,” N. A. Mahynski, R. Mao, E. Pretti, V. K. Shen, and J. Mittal, Soft Matter, 16 3187 (2020).