
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
The National Institute of Standards and Technology is developing a mass spectral library of all identifiable components derived from the digestion of monoclonal antibodies, including peptides, glycans, and glycopeptides. This evaluated tandem mass spectral library aims to provide reference data for laboratories using mass spectrometry to discover and identify monoclonal antibody structures and modifications through the fragmentation of their ions generated by electrospray ionization. It is an extension of the NIST/EPA/NIH Mass Spectral Library and the NIST Peptide Mass Spectral Libraries. Using mass spectral libraries to identify these compounds is more sensitive and robust than interpreting the mass spectra by theoretical methods. These databases are available for testing and integration with existing instrumentation (peptides: https://chemdata.nist.gov/dokuwiki/doku.php?id=peptidew:start, glycans: https://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:msms).
Description
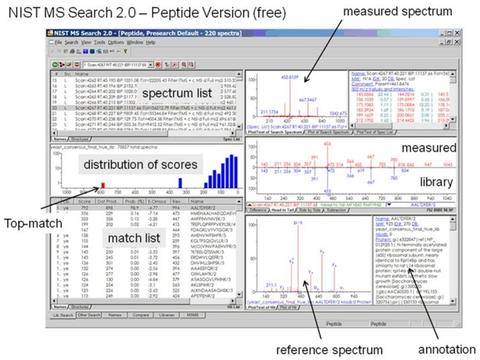
Intended Impact
Modern mass spectrometers used in the field of proteomics and glycomics are capable of profiling hundreds, and even thousands of molecules in a single experiment. Each of these compounds is isolated and fragmented to form a mass spectrum. Therefore, interpretation of these mass spectra is a critical step in the experimental workflow. Since peptide and glycan mass spectra represent physical properties of these molecules, standard interpretation of these mass spectra has the potential to improve the success rate of all discovery experiments in proteomics and glycomics.
Objective
Biological mass spectrometry is a critical tool in understanding monoclonal antibodies. NIST researchers are using their expertise in building mass spectral libraries for other small molecules to compile a comprehensive library of consensus mass spectra of peptides, glycans, glycopeptides, and other important compounds. Developing a standard method for interpreting these mass spectral data and a comprehensive library of high quality tandem mass spectra is critical for establishing and advancing this technology.
Goals
- Develop a mass spectral library of all identifiable components of monoclonal antibodies, including peptide (and its modifications), semi-tryptic, glycan, and glycopeptide ions
- Provide the library in a form that is easily searchable using software tools
- Update and maintain the library as a reference data resource
Research Activities and Technical Approach
| While the mass spectrometers used to identify peptides in proteomics have improved greatly over that past ten years, computer algorithms for peptide identification have not. Traditionally, this process involves a step wherein theoretical peptide fragmentation spectra are predicted from protein sequences. These spectra typically contain peaks at the correct m/z values but contain little or no information about their relative intensities (i.e. peak heights) or less common fragmentation products. |
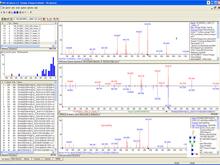
Screenshot of the MS Search 2.0 software showing a library match of an unknown glycan with a glycan in the library. |
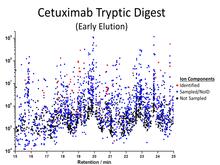
Ion components of a tryptic digest of cetuximab. | Moreover, hundreds of glycans may be detected in a single experiment but the identification of these compounds depends on the availability of libraries of standard reference mass spectra. Mass spectral libraries are built from measured spectra of known compounds and enable the use of sensitive search algorithms. The use of these algorithms and libraries (1) will lead to a higher percentage of identified spectra at the same level of reliability and (2) will greatly increase the robustness of the glycan identification step. |
The data for this project is both being generated 'in-house' at NIST and collected from many outside sources. NIST also has data exchange agreements with several international proteomics data repositories in order to efficiently share the most relevant data.
To date, the small molecule mass spectral library contains >120,000 spectra for >15,000 ions of >7,000 compounds of biological and environmental relevance (including metabolites, bioactive peptides, amino acids and small peptides, sugars and glycans, lipids and phospholipids, drugs, pesticides, surfactants, and various contaminants). Several of the peptide libraries, including human, yeast and E. coli, represent significant coverage of the proteomes and are suitable for routine uses.
The spectra in these libraries have the features of being experimentally validated, critically evaluated, and annotated with great detail. Their use, in combination or as an alternative to sequence-based identification methods, has been shown to double the number of peptide identifications for some data sets.
Major Accomplishments
- Large peptide and glycan mass spectral libraries have been developed for monoclonal antibodies
- A website has been launched for distribution of the libraries and software (peptides: https://chemdata.nist.gov/dokuwiki/doku.php?id=peptidew:start, glycans: https://chemdata.nist.gov/dokuwiki/doku.php?id=chemdata:msms)
- NIST MS Search 2.0 software for peptides and glycans is available
Publications
- Kilpatrick, L.E., Neta, P., Yang, X., Simon-Manso, Y., Liang, Y., Stein, S.E., Formation of y + 10 and y + 11 Ions in the Collision-Induced Dissociation of Peptide Ions, J. Am. Soc. Mass Spectrom., 2012 Apr., 23(4), pp. 655-663.
- Yang, X., Neta, P., Simón-Manso, Y., Kilpatrick, L., Liang, Y., Stein, S. Building a High Quality and Comprehensive Tandem Mass Spectral Library, Poster presented at the 2012 International Conference of the Metabolomics Society.
- Yang, X., Neta, P., Simón-Manso, Y., Liang, Y., Kilpatrick, L., Roth, J., De Leoz, M.L.A., Mi, W., Stein, S.E., Methods for Building High Quality and Comprehensive MS/MS Libraries, Poster presented at the 60th ASMS Conference on Mass Spectrometry and Allied Topics, June 2012.
- De Leoz, M.L.A., Kilpatrick, L.E., Yan, X., Yang, X., Tarlov, M.J., Stein, S.E., Comprehensive Mass Spectral Reference Library of Glycans and Glycopeptides for Therapeutic Antibodies, Talk presented at the 60th ASMS Conference on Mass Spectrometry and Allied Topics, June 2012.
- Rudnick, P.A., Clauser, K.R., Kilpatrick, L.E., Tchekhovskoi, D.V., Neta, P., Blonder, N., Billheimer, D.D., Blackman, R.K., Bunk, D.M., Cardasis, H.L., Ham, A.J., Jaffe, J.D., Kinsinger, C.R., Mesri, M., Neubert, T.A., Schilling, B., Tabb, D.L., Tegeler, T.J., Vega-Montoto, L., Variyath, A.M., Wang, M., Wang, P., Whiteaker, J.R., Zimmerman, L.J., Carr, S.A., Fisher, S.J., Gibson, B.W., Paulovich, A.G., Regnier, F.E., Rodriguez, H., Spiegelman, C., Tempst, P., Liebler, D.C., Stein, S.E., Performance Metrics for Liquid Chromatography-Tandem Mass Spectrometry Systems in Proteomics Analyses, Mol. Cell Proteomics, 2010 Feb., 9(2), pp. 225-241.
- Lam, H., Deutsch, E.W., Eddes, J.S., Eng, J.K., Stein, S.E., Aebersold, R., Building Consensus Spectral Libraries for Peptide Identification in Proteomics, Nat. Methods, 2008 Oct, 5(10), pp. 873-875.
- Lam, H., Deutsch, E.W., Eddes, J.S., Eng, J.K., King, N., Stein, S.E., Aebersold, R., Development and Validation of a Spectral Library Searching Method for Peptide Identification from MS/MS, Proteomics, 2007 Mar., 7(5), pp. 655-657.
Associated Product(s)
NIST Peptide Mass Spectral Libraries for Proteomics and Chemical Analysis
NIST/EPA/NIH Mass Spectral Library with Search Program (SRD 1A) – an interactive access to experimental data produced at NIST and by its customers organized by SRM.
NIST Chemistry Webbook
Electron Ionization Library Component of the NIST/EPA/NIH Mass Spectral Library and NIST GC Retention Index Database
Mass Spectrometry Tools