
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Measurement and Prediction of Local Structure
Summary
Our goal is to provide analytical tools that allow measurement and prediction of local structure to enable the development of ceramic materials for electronic applications. Under this project, we develop data analysis methods for quantitative determination of local structure from multiple experimental techniques, and theoretical methods for prediction of local atomic configurations from first principles.
Description
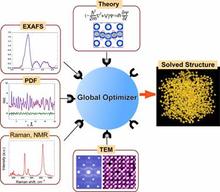
Functional materials enable applications in the computer, data storage, wireless communication, and energy sectors. The properties of many of these systems are critically dependent on deviations of local atomic arrangements from the global average of the crystal. No single measurement technique can provide an accurate description of the local structure. Therefore, our efforts focus on developing analytical tools (methods and software) that allow integration of measurements from multiple experimental methods, and theory to enable accurate and comprehensive determination and prediction of atomic configurations over length-scales from sub-nanometer to macroscopic. Specifically, we develop methods and software for structural refinements using simultaneous fitting of diffraction and x-ray absorption data, and modeling tools for ab initio simulations of diffuse scattering in systems with nanoscale order, Raman spectra in systems with local disorder, which is common for technological materials. All of this input is needed for the determination of atomic order over multiple length scales.
Impact and customers
- The availability of methods and analytical tools for measuring local structure will accelerate the formulation of structure-property relations for advanced functional materials and allow their more rapid selection and optimization, thereby decreasing the development time for new devices.
- Computer software for refinements of atomistic models using simultaneous fitting of X-ray and neutron total scattering data, extended X-ray absorption fine structure, and diffuse scattering in single-crystal X-ray and neutron diffraction is being developed in collaboration with our partners at Spallation Neutron Source (ORNL) and ISIS pulsed neutron and muon source in the United Kingdom. This software package available at https://rmcprofile.ornl.gov has become a standard tool for atomistic structural refinements.
Recent Publications
Levin, I, Yang, F, Maier, R, Laws, WJ, Keeble, DS, Cibin, G, Sinclair, DC, “Displacive Order–Disorder Behavior and Intrinsic Clustering of Lattice Distortions in Bi‐Substituted NaNbO3,” Advanced Functional Materials, 2001840 (2020) https://doi.org/10.1002/adfm.202001840
Krayzman, V, Cockayne, E, Johnston-Peck, AC, Vaughan, G, Zhang, F, Allen, AJ, Kunz, LY, Cargnello, M, Friedman, LH, Levin, I, “Local Structural Distortions and Failure of the Surface-Stress “Core-Shell” Model in Brookite Titania Nanorods,” Chemistry of Materials (2019) https://doi.org/10.1021/acs.chemmater.9b03762
Eremenko, M, Krayzman, V, Bosak, A, Playford, HY, Chapman, KW; Woicik, JC, Ravel, B; Levin, I; “Local atomic order and hierarchical polar nanoregions in a classical relaxor ferroelectric,” Nature communications,10, 1, 1-9, (2019) https://doi.org/10.1038/s41467-019-10665-4
Levin, I, Keeble, DS, Cibin, G, Playford, HY, Eremenko, M, Krayzman, V, Laws, WJ, Reaney, IM, “Nanoscale polar heterogeneities and branching Bi-displacement directions in K0. 5Bi0. 5TiO3,” Chemistry of Materials, 31, 7, 2450-2458 (2019) https://doi.org/10.1021/acs.chemmater.8b05187
Major Accomplishments
Complex perovskite-structured metal oxides form the backbone of advanced electroceramics. In many of these systems, competing interatomic interactions generate local deviations from average periodicity which modify, often critically, a material’s response to an external stimulus (e.g. electric field). Because of their relative subtlety and complexity, the exact nature of these average-structure perturbations remains uncertain even for classical and extensively studied compounds. Relaxor ferroelectrics represent a prominent example of technologically useful materials whose structure remains elusive despite many decades of intense research. As a result, the origin of the relaxor behavior is still not fully understood. Recently, we have been able to elucidate the much-debated nanoscale structure of a classical relaxor ferroelectric PbMg1/3Nb2/3O3 (PMN) by combining neutron and X-ray total scattering measured on powder samples with a 3D distribution of the X-ray diffuse-scattering intensity obtained for a single crystal. This work capitalized on our recent major development of the computer software for atomistic structure refinements, which enabled a consistent description of the atomic arrangements over a broad range of length-scales in a single model, permitting the use of structural models sufficiently large to represent systems, like PMN. Our results clarified several contentious hypotheses regarding the chemical and polar ordering in PMN and revealed a hitherto unsuspected hierarchical nature of polar nanoregions that are key to the relaxor properties.

Nanoparticles are another example of technological systems where traditional methods of structure determination which rely on Bragg diffraction peaks become ineffective. Titania nanoparticles attract interest for their potentially useful photocatalytic properties. In bulk titania crystals under ambient conditions, rutile is the thermodynamically stable phase, whereas in nanoparticles, anatase and brookite become preferred. Commercial adaptation of TiO2 in photocatalysts is hindered by its wider-than-optimal band gap and fast electron-hole recombination after absorption of light. These issues can be mitigated by engineering nanoparticles with specific shapes, dimensions, and defect populations. Indeed, promising photocatalytic properties have been reported for nanoparticles of all three polymorphs and mixtures thereof. However, the exact mechanism of the improved performance in each case remains uncertain. In nanoparticles, the already distinct electronic structures of bulk polymorphs are modified because of structural relaxations induced by particle-size and surface effects. The effects of structural distortions, which can vary between the particle’s interior and surface, on electronic properties relevant to photocatalytic performance are uncertain, in part because such distortions themselves have not yet been characterized in detail. We demonstrated structural refinements of whole-nanoparticle atomistic models for TiO2 brookite nanorods, which exhibit enhanced photocatalytic properties. Our results revealed that the atomic positions in the nanorods differ from the bulk structure by static atomic displacements that increase progressively from the nanorod axis toward its side surfaces. A pattern of structural relaxations exhibited symmetry consistent with that imposed by the nanorod habits. Density-functional-theory calculations were in qualitative agreement with the experimental observations and additionally revealed a strong dependence of the structural relaxations, including lattice-volume changes, on sorbate coverage. The experimental and computed magnitudes of these relaxations appeared to be significantly larger than those predicted by a simple surface-stress model, with the difference attributed to electrostatic interactions. This study demonstrated the potential of using atomistic structure refinements to obtain a detailed picture of atomic order in nanoparticles, and highlighted complex, spatially variable distortions that need to be considered while interpreting the electronic properties of these materials.

Associated Product(s)
Project Summary (PDF)
Software
Utility programs for RMCProfile and documentation for use are available for download
Bayesian denoising for continuous signals