
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
LAMMPS MD: Equation of State (pressure vs. density) - TraPPE Carbon Dioxide
The main purpose of the following data set is to present equation of state (density-pressure-temperature) data for a version of the TraPPE Carbon Dioxide fluid that was obtained using the LAMMPS Molecular Dynamics (MD) simulation suite. The secondary purpose of this data set is to provide sample LAMMPS input and initial configuration files that an end user may use in LAMMPS to obtain the same equation of state data, to either verify output from an installation of LAMMPS or educate the user about the basic features of LAMMPS. We first present output from LAMMPS, and compare it to Monte-Carlo derived results, and then provide a tutorial on how to reproduce that same data using LAMMPS.
Simulation details
| Simulation Package | LAMMPS - see installation tutorial |
| Method | Canonical Ensemble (fixed N, V, T) Molecular Dynamics using the LAMMPS "rigid/nvt" ensemble option, which includes constraints to preserve bond lengths and angles |
| Number of TraPPE Carbon Dioxide Molecules | 1000 |
| Simulation Cell | Cubic cell, volume set with constant N=1000 to achieve desired reduced density |
| Truncation | Lennard-Jones Dispersion: 15 Å + analytic Long-range Corrections; Electrostatics: 15 Å + Long-range correction [Particle-particle particle-Mesh (PPPM) Ewald] |
| Time Step | 1.0 fs |
| Thermostat | Temperature imposed via Nosé-Hoover chained thermostats (three, LAMMPS default) with Tdamp=100 fs |
| Simulation length | 1.1x106 time steps (intended as 105 equilibration steps followed by 106 production steps) |
Additional simulation details are in the sample input file, see below.
Results
1. Results for select temperatures (tabular)
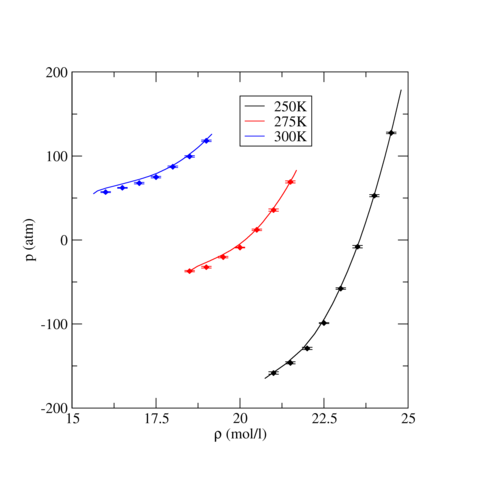
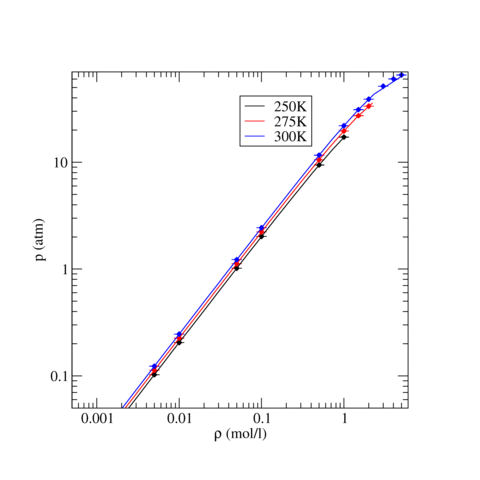
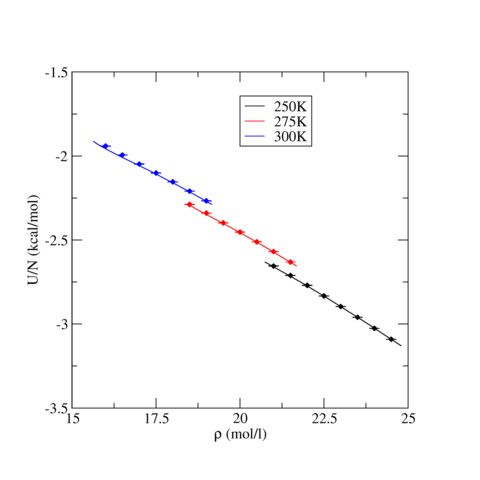
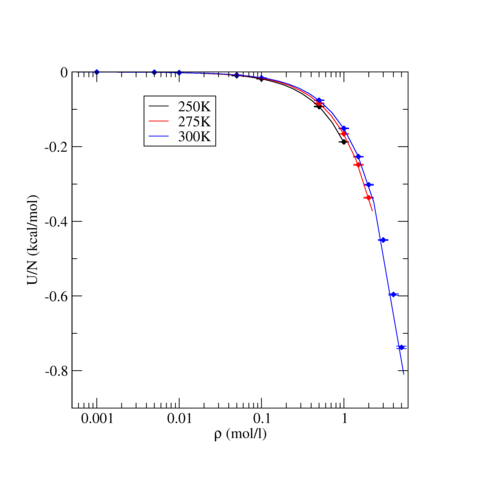
The following table contains pressure and intermolecular potential energy per TraPPE Carbon Dioxide molecule as a function of density for three temperatures, T=250, 275, and 300 K. The ensemble averages were calculated the LAMMPS runs described above and uncertainty, stated by the standard error, were estimated using block analysis (see below). For all three temperatures, phase stability is noted if applicable.
Liquid State Points
| T = 250 K | T = 275 K | T = 300 K | ||||||||||
| ρ (mol/l) | p (atm) | +/- | U/N (kcal/mol) | +/- | p (atm) | +/- | U/N (kcal/mol) | +/- | p (atm) | +/- | U/N (kcal/mol) | +/- |
| 16.0 | unstable | unstable | 5.700E+01 | 5.966E-01 | -1.941E+00 | 1.272E-03 | ||||||
| 16.5 | unstable | unstable | 6.213E+01 | 4.923E-01 | -1.994E+00 | 8.625E-04 | ||||||
| 17.0 | unstable | unstable | 6.766E+01 | 8.585E-01 | -2.048E+00 | 4.515E-03 | ||||||
| 17.5 | unstable | unstable | 7.485E+01 | 8.420E-01 | -2.101E+00 | 1.371E-03 | ||||||
| 18.0 | unstable | unstable | 8.721E+01 | 9.592E-01 | -2.154E+00 | 5.541E-04 | ||||||
| 18.5 | unstable | -3.711E+01 | 7.899E-01 | -2.288E+00 | 1.246E-03 | 9.950E+01 | 9.286E-01 | -2.209E+00 | 4.145E-04 | |||
| 19.0 | unstable | -3.239E+01 | 9.072E-01 | -2.340E+00 | 7.605E-04 | 1.181E+02 | 1.003E+00 | -2.267E+00 | 6.690E-04 | |||
| 19.5 | unstable | -2.043E+01 | 9.185E-01 | -2.398E+00 | 8.949E-04 | did not simulate | ||||||
| 20.0 | unstable | -8.996E+00 | 4.819E-01 | -2.453E+00 | 8.576E-04 | did not simulate | ||||||
| 20.5 | unstable | 1.206E+01 | 8.714E-01 | -2.511E+00 | 7.128E-04 | did not simulate | ||||||
| 21.0 | -1.584E+02 | 1.435E+00 | -2.655E+00 | 1.042E-03 | 3.566E+01 | 1.345E+00 | -2.569E+00 | 4.982E-04 | did not simulate | |||
| 21.5 | -1.463E+02 | 1.254E+00 | -2.711E+00 | 7.017E-04 | 6.911E+01 | 1.232E+00 | -2.631E+00 | 4.663E-04 | did not simulate | |||
| 22.0 | -1.291E+02 | 1.311E+00 | -2.770E+00 |
6.590E-04 |
did not simulate | did not simulate | ||||||
| 22.5 | -9.887E+01 | 5.425E-01 | -2.833E+00 | 5.784E-04 | did not simulate | did not simulate | ||||||
| 23.0 | -5.780E+01 | 8.110E-01 | -2.896E+00 | 4.864E-04 | did not simulate | did not simulate | ||||||
| 23.5 | -7.985E+00 | 1.440E+00 | -2.960E+00 | 3.978E-04 | did not simulate | did not simulate | ||||||
| 24.0 | 5.277E+01 | 1.137E+00 | -3.026E+00 | 3.042E-04 | did not simulate | did not simulate | ||||||
| 24.5 |
1.275E+02 |
8.420E-01 | -3.091E+00 | 2.018E-04 | did not simulate | did not simulate | ||||||
Gas State Points
| T = 250 K | T = 275 K | T = 300 K | ||||||||||
| ρ (mol/l) | p (atm) | +/- | U/N (kcal/mol) | +/- | p (atm) | +/- | U/N (kcal/mol) | +/- | p (atm) | +/- | U/N (kcal/mol) | +/- |
| 0.001 | 2.051E-02 | 6.331E-06 | -1.976E-04 | 2.595E-05 | 2.257E-02 | 1.206E-05 | -1.777E-04 | 2.758E-05 | 2.460E-02 | 6.180E-06 | -1.828E-04 | 3.574E-05 |
| 0.005 | 1.025E-01 | 3.588E-05 | -8.233E-04 | 5.025E-05 | 1.128E-01 | 2.563E-05 | -8.597E-04 | 3.985E-05 | 1.231E-01 | 4.454E-05 | -7.953E-04 | 3.469-05 |
| 0.01 | 2.048E-01 | 7.184E-05 | -1.948E-03 | 1.176E-04 | 2.254E-01 | 4.619E-05 | -1.729E-03 | 9.418E-05 | 2.458E-01 | 4.849E-05 | -1.851E-03 | 9.660E-05 |
| 0.05 | 1.017E+00 | 2.137E-04 | -1.005E-02 | 2.113E-04 | 1.121E+00 | 2.056E-04 | -7.711E-03 | 8.644E-05 | 1.225E+00 | 4.005E-04 | -8.231E-03 | 2.619E-04 |
| 0.1 | 2.018E+00 | 6.699E-04 | -1.850E-02 | 3.458E-04 | 2.228E+00 | 4.560E-04 | -1.667E-02 | 2.554E-04 | 2.435E+00 | 3.171E-04 | -1.572E-02 | 1.763E-04 |
| 0.5 | 9.423E+00 | 7.792E-03 | -9.249E-02 | 1.031E-03 | 1.053E+01 | 7.682E-03 | -8.391E-02 | 8.653E-04 | 1.165E+01 | 7.389E-03 | -7.573E-02 | 7.147E-03 |
| 1.0 | 1.719E+01 | 1.050E-02 | -1.873E-01 | 6.225E-04 | 1.962E+01 | 1.337E-02 | -1.656E-01 | 5.899E-04 | 2.198E+01 | 1.909E-02 | -1.510E-01 | 9.237E-04 |
| 1.5 | unstable | 2.730E+01 | 3.379E-02 | -2.485E-01 | 1.088E-03 | 3.110E+01 | 3.098E-02 | -2.267E-01 | 7.936E-04 | |||
| 2.0 | unstable | 3.350E+01 | 4.241E-02 | -3.367E-01 | 1.263E-03 | 3.895E+01 | 5.600E-02 | -3.020E-01 | 1.323E-03 | |||
| 3.0 | unstable | unstable | 5.155E+01 | 5.681E-02 | -4.500E-01 | 1.184E-03 | ||||||
| 4.0 | unstable | unstable | 6.027E+01 | 8.008E-02 | -5.959E-01 | 1.729E-03 | ||||||
| 5.0 | unstable | unstable | 6.566E+01 | 1.854E-01 | -7.376E-01 | 3.307E-03 | ||||||
2. Pressure-density-energy equation of state
The following graphics show the pressure-density equations of state and the internal energy per molecule versus density in the form of a phase diagram for select temperatures (250, 275, and 300 K). In both graphics, the solid lines are the corresponding equation of state and energy as calculated from Grand Canonical-Transition Matrix Monte Carlo Simulations (GC-TMMC, see results elsewhere in the NIST SRSW). Solid symbols indicate the results from LAMMPS simulations, with the standard error shown by error bars. There is good agreement between the two sets of results, particularly when considering that significantly different system sizes were used (N=1000 for MD versus N<500 for MC). Some deviation between between the two methods is expected for the higher temperatures, which approach the critical temperature of TraPPE Carbon Dioxide.
Reproducing output
The LAMMPS MD results in the preceding table and graphics may be reproduced using example LAMMPS runs described as follows.
1. LAMMPS Installation
The data shown above were generated using a generic installation of LAMMPS, the executables of which may be reproduced as described here. This tutorial assumes some level of familiarity with POSIX-compliant operating systems (e.g., Unix, Linux, or Mac OSX) at the command-prompt level and access to a system with the GCC compiler, OpenMPI parallelization suite, Python, and the git version control system. This tutorial uses ">" to indicate the shell command prompt and $LAMMPS_DIR to identify the directory where LAMMPS is downloaded and later compiled. First, LAMMPS can be obtained using the instructions at http://lammps.sandia.gov/download.html#git via:
>git clone https://github.com/lammps/lammps.git
Second, LAMMPS executables may be compiled via:
>cd $LAMMPS_DIR
>git checkout stable_31Mar2017
>cd src
>make purge
>make package-update
>make yes-user-misc yes-molecule yes-rigid yes-kspace
>make -j8 mpi
The installation sequence 1) switches to the "stable_31Mar2017" commit of LAMMPS (to ensure that a user is using the same code used to generate the data shown above), 2) removes any existing installation of LAMMPS, 3) updates any out of date packages, 4) installs the "USER-MISC", "MOLECULE", "RIGID", and "KSPACE" packages to enable the simulation of a rigid molecule using Ewald-type long-range electrostatics, and 5) builds an MPI-enabled executable using eight processor cores for parallel compilation. The end result is an executable named "lmp_mpi" located in the $LAMMPS_DIR/src directory.
Finally, we used the following operating system, compiler, and MPI libraries to build the LAMMPS executable:
Linux OS: CentOS 7; 3.10.0-514.21.1.el7.x86_64 kernel
GCC: v4.8.5 (2015-06-23, listed as Red Hat 4.8.5-11)
OpenMPI: v1.10.3 (2016-06-14)
LAMMPS: Git Checkout Tag stable_31Mar2017 (2017-03-31)
2. Example LAMMPS Scripts
Example LAMMPS scripts and initial configurations that will yield the data shown above may be obtained from a git repository at: https://github.com/dwsideriusNIST/LAMMPS_Examples, e.g. via:
>git clone git [at] github.com:dwsideriusNIST/LAMMPS_Examples.git
This tutorial assumes that the git repository resides in $EXAMPLES_DIR. The relevant inner directories are:
| TraPPECO2_initial_cfgs |
Initial configurations of N=1000 TraPPE Carbon Dioxide molecules at the specified density |
| run_scripts | Relevant LAMMPS Scripts |
| analysis | Python scripts to post-process LAMMPS output |
| TraPPECO2_example | Example shell script that will run a LAMMPS simulation at ρ=18.0 mol/l |
3. Example LAMMPS Run
A short example LAMMPS run is available in $EXAMPLES_DIR/TraPPECO2_example, which may be run to confirm that LAMMPS is operating properly. This simulation is executed via:
>cd $EXAMPLES_DIR/TraPPECO2_example
>sh example.sh
In the "example.sh" script, one can see that this script runs an TraPPE Carbon Dioxide simulation at ρ=18.0 mol/l and T=300 K for 60000 time steps (all LAMMPS settings are as shown previously, using the Particle-particle Particle-mesh k-space option) then analyzes the MD trajectory to report the average temperature, pressure, and potential energy. The final output, from the block analysis script, will report the following ensemble averages and uncertainty:
Thermodynamic Ensemble Averages from: ave.dens_18.0molL.out
Discarded Timesteps: 10000
Number of Blocks: 5
Block size (timesteps): 10000
Stated uncertainty is the standard error of the thermodynamic property
Temperature +3.0008E+02 +/- +1.3188E-01
Density +7.9219E-01 +/- +0.0000E+00
PotentialEnergy -2.1523E+03 +/- +4.7930E+00
Pressure +8.0869E+01 +/- +2.5806E+00
We note that the average pressure and energy differ from those in the table above because this example run was only 60000 timesteps.
4. Production LAMMPS Runs
>mpirun -np $NP lmp_mpi -in TraPPECO2.NVT -var infile $FOO -var temp $BAR >$EXAMPLES_DIR/analysis/block_analysis.py -f $FILENAME -b $BLOCKS -m $STEPS_SKIP >cd $EXAMPLES_DIR >mkdir test ; cd test >cp ../run_scripts/TraPPECO2.NVT ./ >cp ../TraPPECO2_initial_cfgs/TraPPECO2_N1000_config.dens_18.0molL.cfg.lammps ./ >mpirun -np 12 lmp_mpi -in TraPPECO2.NVT -var infile TraPPECO2_N1000_config.dens_18.0molL.cfg.lammps -var temp 300.0 >../analysis/block_analysis.py -f ave.out -b 10 -m 100000Thermodynamic Ensemble Averages from: ave.out
Discarded Timesteps: 100000
Number of Blocks: 10
Block size (timesteps): 100000
Stated uncertainty is the standard error of the thermodynamic property
Temperature +3.0017E+02 +/- +3.4814E-02
Density +7.9219E-01 +/- +0.0000E+00
PotentialEnergy -2.1537E+03 +/- +5.5411E-01
Pressure +8.7210E+01 +/- +9.5917E+00