
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Machine Learning Algorithm Development and Artificial Intelligence Benchmarking
Summary
Using AI tools and physics-, chemistry-, and materials science-based knowledge, this project is aimed at developing machine learning (ML) algorithms to enable rapid and accurate selection of optimal system features and performance, as well as material discovery, under a variety of conditions. Particular attention is paid to uncertainty quantification and model robustness.
Description
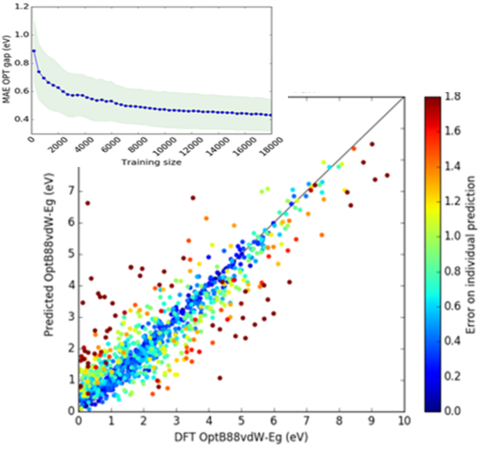
- Atomistic machine learning — Machine Learning is an important means of accelerating prediction of atomistic properties of crystalline solids and molecules. All models are trained and validated using density functional theory (DFT) data from the NIST JARVIS project (https://jarvis.nist.gov/). Formation energies, electronic band gaps, static refractive indices, magnetic properties, and modulus of elasticity for three-dimensional materials as well as exfoliation energies of two-dimensional (2D)-layered materials are among the properties these ML models have proven successful at predicting. Spectral properties, like phonon density of states, are among more complex material characteristics that our algorithms can evaluate.
- CFID models are characterized by the use of a complete set of chemo-structural descriptors (pairwise radial, nearest-neighbor, bond-angle, dihedral-angle, and core-charge distributions, in addition to standard chemical descriptors), which allows differentiating between structural prototypes and quickly predicting a variety of material properties. Learn more about JARVIS-ML.
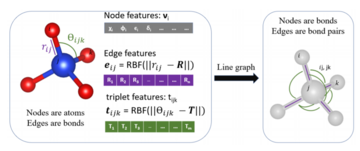
- Atomistic Line Graph Neural Network (ALIGNN) is a graph neural network architecture that explicitly incorporates bond angles, which are critical for distinguishing many atomic structures. We developed ALIGNN models for predicting 52 solid-state and molecular properties available in the JARVIS-DFT, Materials project, and QM9 databases. We also developed a unified atomistic line graph neural network-based force field (ALIGNN-FF) that can model both structurally and chemically diverse solids with any combination of 89 elements from the periodic table. Tools for training and deploying ALIGNN models are available at https://github.com/usnistgov/alignn. Read more about JARVIS-ALIGNN and JARVIS-ALIGNN-FF.
- AtomVision — AtomVision is a machine vision library for atomistic images that can be used to generate and curate microscopy image (such as scanning tunneling microscopy and scanning transmission electron microscopy) data sets and apply a variety of machine learning techniques. The AtomVision library is available at https://github.com/usnistgov/atomvision.
- Uncertainty and robustness characterization for atomistic ML models –
- Research into uncertainty quantification and calibration for atomistic machine learning systems
- We also investigate the data and training factors underlying the generalization performance of ML models
- Leveraging Theory for Enhanced Machine Learning — The application of machine learning to the materials domain has traditionally struggled with two major challenges: a lack of large, curated data sets and the need to understand the physics behind the machine-learning prediction. The former problem is particularly acute in the polymers domain. Here we aim to simultaneously tackle these challenges through the incorporation of scientific knowledge, thus, providing improved predictions for smaller data sets, both under interpolation and extrapolation, and a degree of explainability.
Major Accomplishments
- Co-organized Artificial Intelligence for Materials Science (AIMS) workshops (https://jarvis.nist.gov/events/aims) in 2018, 2019, 2022 (https://www.nist.gov/news-events/events/2022/07/artificial-intelligence-materials-science-aims), 2023 (upcoming https://www.nist.gov/news-events/events/2023/07/artificial-intelligence-materials-science-aims-workshop)
- Creation of GitHub Repository for AI codes and tools associated with the Atomistic ML project (as part of the larger JARVIS project) (https://github.com/usnistgov/jarvis/tree/master/jarvis/ai)