Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
JARVIS-ALIGNN, JARVIS-ALIGNN-FF
Summary
Our Atomistic Line Graph Neural Network (ALIGNN) models are designed to predict atomistic properties with high accuracy. They can successfully predict single value, spectral and gradient based properties. There are more than 100 properties of materials that have been trained using this model. Some of the single value ones are formation energy, electronic band gap, bulk and shear moduli, dielectric constants, magnetic moment, Seebeck coefficient, and power factor. Spectra well captured by ALIGNN include phonon density of states, electron density of states, Eliashberg function, infrared and Raman.
To model larger scale systems and predict gradient properties such as forces, we develop a unified atomistic line graph neural network-based force field (ALIGNN-FF) that can model both structurally and chemically diverse solids with any combination of 89 elements from the periodic table.
Description

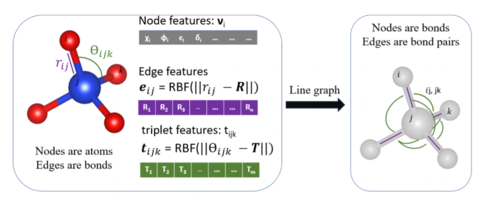
ALIGNN uses a line graph neural networks to include bond distances and angular information graph to incorporate finer details of atomic structure, leading to high accuracy models. While the nodes of an atomistic graph correspond to atoms and its edges correspond to bonds, the nodes of an atomistic line graph correspond to interatomic bonds and its edges correspond to bond angles. Our model alternates between graph convolution on these two graphs, propagating bond angle information through interatomic bond representations to the atom-wise representations and vice versa.
ALIGNN is a part of the Joint Automated Repository for Various Integrated Simulations (JARVIS) infrastructure. We train ALIGNN models for several crystalline material properties from JARVIS-density functional theory (JARVIS-DFT) and Materials project (MP) datasets as well as molecular properties from QM9 database.
Large scale atomistic simulation of multi-component systems is a difficult task, but they are highly valuable for industrial applications such as designing alloys, designing electrical contacts, touch screens, transistors, batteries, composites, and catalysts. Classical force-fields (FF), or interatomic potentials, can be used for such simulations but they are usually parameterized for a very narrow chemical phase-space limiting their applicability and transferability. We extend the ALIGNN model to also predict derivatives that are necessary for FF formalism. This way, we were able to develop a unified atomistic line graph neural network-based force field (ALIGNN-FF) that can model both structurally and chemically diverse solids with any combination of 89 elements from the periodic table.