
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Fundamental Studies of the Conformational Phase Behavior of Chain Molecules at Interfaces
Summary
Chain molecules tethered or adsorbed to solid surfaces are at the heart of a wide range of technologies. Examples include colloidal particle stabilization, chromatographic materials, and drug delivery methods. Molecular dynamics simulations of covalently modified surfaces provide models of conformational details that depend on chain length, chain density, and temperature. These models offer insight into selectivity trends observed in liquid chromatography with C18 and C30 stationary phases, prepared by monomeric or polymeric synthetic approaches, and operated at different column temperatures. Conformational details of tethered alkyl chains have also been studied using advanced Monte Carlo simulation techniques that provide the free energy of the system. In this effort, we systematically determine how factors such as chain length, chain density, and chain-surface interactions impact the resulting structure of adsorbed and tethered polymers.
Description
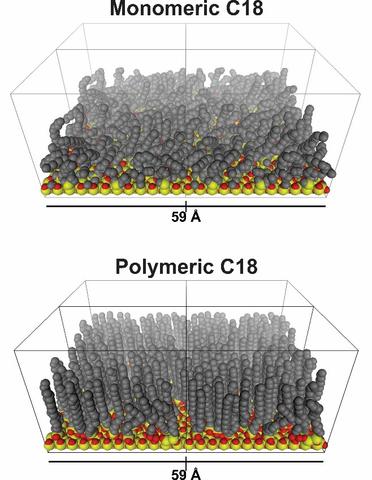
Representation of octadecyl stationary phase morphology showing an ordered state and a disordered state.
Intended Impact
Predicting how the conformational phase behavior of tethered chain molecules depends on external variables is challenging. Understanding how even simple models for tethered chain molecules depend on external variables will greatly aid our understanding of tethered chain systems.
Goals
To determine how variables such as temperature, chain density, chain length, surface interactions, etc., impact the conformational phase behavior of tethered chains.
Research Activities
We have investigated the conformational phase behavior of tethered chain systems using temperature expanded Monte Carlo techniques at thousands of state points, covering densities from the isolated chains to the thin film limit, and a broad range of temperatures. To date, we have investigated the impact of surface interactions, chain stiffness, and tethering geometry on conformational phase behavior.
In a separate effort, alkyl stationary phases used in liquid chromatography have been modeled by molecular dynamic simulations, under chromatographically relevant conditions. Stationary phase order was found to increase with increased bonding density, increased chain length, and at decreased temperature.
Major Accomplishments
We have demonstrated that simulation may be used as an effective guide to understanding the conformational phase behavior of tethered chain systems.
Publications
1. Sander, L. C., "Separations by Shape: Molecular Shape Recognition in Liquid Chromatography," Chromatographia, 85, 299-305 (2022).
2. Lippa, K. A. and Sander, L. C., "Identification of isolated cavity features within molecular dynamics simulated chromatographic surfaces," Journal of Chromatography A, 1128, 79-89 (2006).
3. Lippa, K. A., Sander, L. C., and Mountain, R. D., "Molecular dynamics simulations of alkylsilane stationary-phase order and disorder. 1. Effects of surface coverage and bonding chemistry," Analytical Chemistry, 77, 7852-7861 (2005).
4. Lippa, K. A., Sander, L. C., and Mountain, R. D., "Molecular dynamics Simulations of alkylsilane stationary-phase order and disorder. 2. Effects of temperature and chain length," Analytical Chemistry, 77, 7862-7871 (2005).
5. Sander, L. C., Lippa, K. A., and Wise, S. A., "Order and disorder in alkyl stationary phases," Analytical and Bioanalytical Chemistry, 382, 646-668 (2005).