
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Atomistic tools for structure-property investigations
Summary
This project provides and develops novel computational infrastructure (i.e. data archives, open-source codes, APIs) that support the design, execution, and analysis of classical atomistic calculations. This includes the following activities:
-
Developing and maintaining the public Interatomic Potentials Repository that includes a database of interatomic potentials and associated property calculations to assist users in accessing and selecting between existing potentials.
-
Providing tools to support the development of new interatomic potentials and the generation and analysis of atomistic structures.
-
Developing a high throughput calculation infrastructure that uses reproducible atomistic calculation methods.
-
Evaluating crystal and crystalline defect properties in high throughput across different interatomic potentials.
Description
Interatomic Potential Repository

The Interatomic Potentials Repository (IPR) provides a source for interatomic potentials (force fields), related files, and evaluation tools to help researchers obtain interatomic models and judge their quality and applicability. The files provided are of known provenance and have either been submitted or vetted by their developers or were obtained from other trusted databases. Interatomic potentials and/or related files are currently available for various metals, semiconductors, oxides, and carbon-containing systems.
All content found on the Interatomic Potentials Repository is included in the CDCS database hosted at potentials.nist.gov. The content can be accessed, explored, and downloaded by going to potentials.nist.gov, or by using the potentials Python package available on Github. The potentials package also includes extra tools supporting the use and construction of the hosted parameter files.
Atomistic Manipulation Toolkit (atomman)
The atomman Python package contains tools for constructing and analyzing atomic configurations, with a focus on crystalline defects. It is meant to facilitate the rapid design and development of simulations that are fully documented and easily adaptable to new potentials, configurations, etc. The underlying configuration representation is made general to better support both small- and large-scale configurations, with conversions to many known formats.
iprPy High-Throughput Framework
The iprPy Python package collects complete atomistic calculation methods for performing the property evaluations hosted by the Interatomic Potentials Repository and contains tools for running high-throughput workflows of those calculations. The included calculations are designed with a focus on reproducibility and transparency of the methods.
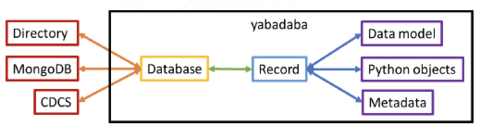
yabadaba python package
The yabadaba python package provides a mid-level abstraction of databases and database records that allows for the construction of user-friendly database APIs. Database interactions are abstracted across multiple database infrastructures, while record content is abstracted by defining schema-specific transformations and interpretations. In this way, APIs built using yabadaba allow for end users to easily query, explore, copy, and interact with the contents of a database without requiring much knowledge of the database infrastructure itself.
JARVIS-FF
The JARVIS-FF (force fields) database is a collection of LAMMPS calculation-based data covering crystal structure, formation energy, phonon density of states, band structure, surface energy and defect formation energy. It is designed to help selecting the optimal FF for the user’s application. Currently the database contains ~110 FFs and ~1500 materials. For each material, the initial crystal structures are obtained from the JARVIS-DFT database, then automatically converted into LAMMPS format inputs and optimized, before performing the LAMMPS calculations to produce the afore mentioned properties. When possible, these properties are compared to corresponding DFT data, to help evaluate the quality of the force-fields for a specific application
Physically-informed Neural Network (PINN) potentials

PINN potentials represent a novel method for describing bonding forces between atoms. The method combines the transferability of physically derived analytic models with the flexibility and accuracy of neural networks. Currently efforts are underway at NIST to develop PINN potentials for several chemical systems including Si, Cu, Pt, Al, Ge, and Ta.
PYFIT-FF
PYFIT-FF is a python package for training artificial neural network interatomic potentials (including PINN potentials). This is done using the PyTorch optimization library which is highly optimized for accelerated computation via CPU or GPU hyper-threading. The main benefits of PYFIT is that it is simple, highly portable, computationally efficient, flexible, well documented, and open source.