
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
Density functional theory (DFT) is the workhorse of modern quantum mechanics calculations of molecular and periodic structures. Countless studies have demonstrated the accuracy and applicability of DFT to a wide variety of systems. Though numerous validation studies have been carried out, few of these have targeted the types of industrially-relevant, materials-oriented systems that will be the subject of the present study. The goal of this study is to address questions such as which functional should I use for this calculation? how large can I expect the deviation from the experimental value to be? which pseudopotential yields the best results? how will the same calculations using different functionals or pseudopotential basis sets differ? on what systems do certain functionals fail? on which do they do particularly well?
Description
A variety of systems will be considered in this work:
- Pure and alloy solids will be studied using periodic DFT methods with three distinct crystal structures that are relevant to the widely-used CALPHAD (Computer Coupling of Phase Diagrams and Thermochemistry) method. This study emphasizes the limits of current codes with respect to convergence in their input parameters and further examines the agreement and disagreement between different codes, functionals, and pseudopotentials. A pilot study is underway for Si, and additional metallic systems will be considered in the future.
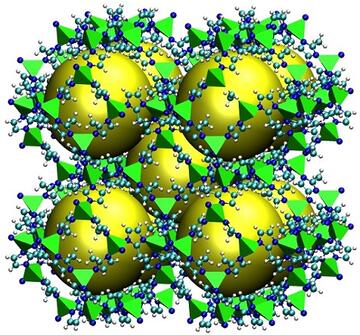
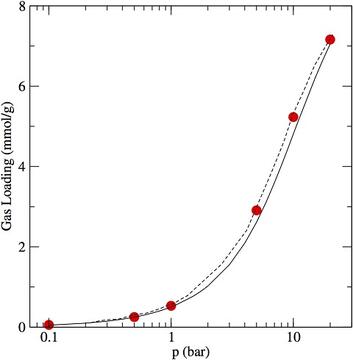
- One of the the crucial aspects in high-throughput computational screening of metal-organic frameworks (MOFs) for carbon capture and other applications is the set of partial charges used in the computations. Here, we focus on the partial charges of the MOF framework atoms. Recently we have begun comparing partial charges derived from several partial charge schemes, as well as studying the effect of the choice of functional and pseudopotential basis set on the derived partial charges. These partial charges are used in Flat-Histogram Monte Carlo codes to calculate adsorption properties, which may be directly compared to experimental measurements being conducted at NIST. Further comparison of the optimized MOF geometry to experimental determinations may be made, and lend some support to the validity of the DFT approach being used. Comparison among different partial charge methods does, however, indicate that equilibrium properties of the MOF-adsorbate system depend heavily on the type of charge calculation scheme. This illustrates the need for particular caution when using so-called "transferable force fields" to model the sorbent properties of MOFs.
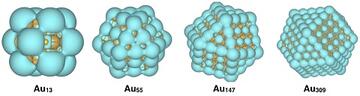
- Metallic nanoparticles (NPs) have found a wide variety of applications in a number of industries and are the subject of intense research. Recent work has focused on determining the reactivity of various transition metal NPs used as fuel cell catalysts via the methods of Chemical Reactivity Theory (CRT). Building on this work, we will compare the results from different DFT functionals and pseudopotentials for a variety of NP properties such as geometry, vibrational frequencies of surface-bound ligands, and optical and magnetic properties. These results will be compared to experimental results wherever possible. As a first step, well-characterized noble-metal NP's will be studied by the above-mentioned strategy. We will subsequently select a variety of industrially-relevant materials with the expectation that the results will be of immediate use in helping industry choose the best methods for their calculations.
Results from these studies will be disseminated via web databases. In particular, we will leverage the infrastructure built to support the popular NIST Computational Chemistry Comparison and Benchmark Database (CCCBDB) to share our work.