
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
TMMC: Reference Adsorption Isotherms for N2 in Metal-organic Frameworks
Grand-canonical Transition-Matrix Monte Carlo (GC-TMMC) simulations [1, 5-10] of N2 were performed at T = 300 K and 350 K in three metal-organic frameworks (MOFs). The particle number range was divided into windows advanced trial moves were performed to ensure adequate sampling at high densities. The main result of a GC-TMMC simulation is the particle number probability distribution (PNPD), which is constructed by stitching together the particle number distributions from each window. The adsorption isotherm may be determined from the PNPD [10]. At each value of N, average total potential energies U were collected and these quantities were used to compute the isosteric heat of adsorption.
Simulations used different combinations of Monte Carlo moves depending on the number of particles, as described below. Low density windows used a conventional set of moves (i.e., no configurational bias) whereas high density windows used a configurational bias strategy known as Dual-cut configurational bias (DCCB) Monte Carlo [13]. Additionally, low-density windows started in Wang-Landau biasing mode to quickly generate a guess of the PNPD, later switching to TMMC mode; data from the TMMC phase of the simulation was saved and reported here.
All simulations were performed using the open-source FEASST Monte Carlo engine [14], version v0.21.1 (commit hash fad5d0d6575a2ed8251d7093e1a250d634cbbcb4).
Other key simulation details common to all simulations are given below:
| Fluid Model | TraPPE N2[11] | |
| Lennard-Jones cutoff | 12A, with linear-force shift tail correction | |
| Ewald Parameters | Set according to DL_POLY recipe [15], with relative tolerance 10-5 | |
| Standard Move Set (N < 100) | Translation, weight 0.3 Rotation, weight 0.2 Insertion/Deletion, weight 0.4 | |
| Advanced Move Set (N ≥ 100) | Translation, weight 0.1 DCCB Regrowth (single Nitrogen), weight 0.06 DCCB Regrowth (both Nitrogens), weight 0.04 DCCB Growth (full N2 particle), weight 0.8 | |
| DCCB Details | Number of trials per atom: 4 Reference Potential: Lennard-Jones + Ewald Realspace Cutoff Radius for Reference Potential: 6A | |
| Total MC Trials | 1.0e8 | |
| Bias update freq | 1.0e4 | |
| Physical Parameters | CODATA 2018 [12] | |
The adsorbent MOFs were reconstructed from publicly-available crystal structures and replicated to ensure that the simulation cell was at least twice the cutoff radius in all dimensions. Forcefields for each MOF were taken from published literature. The MOF structure and forcefield are provided in FEASST particle files in the data repository associated with this page (see "Data Availability" below). Coulombic interactions were handled using the Ewald summation method [2,3] (parameters listed in the metadata files). Lorentz-Berthelot mixing rules were used to set the unlike-atom Lennard-Jones parameters.
Simulation details specific to each MOF
| ZIF-8 | CuBTC | IRMOF-1 | |
| Number of Windows | 60 | 40 | 40 |
| Nmax | 350 | 245 | 275 |
| Unit Cell Replication (Nx, Ny, Nz) | (2,2,2) | (1,1,1) | (1,1,1) |
| Cubic Box Dimensions (A) | 34.023240 | 26.3430 | 25.6690 |
| Simulation MOF Mass (amu) | 21846.91 | 9677.91 | 6158.94 |
| MOF Forcefield, Reference | Snurr [16] | Calero IV [17] | DREIDING [19] + m-CBAC [20] |
| FEASST MOF Particle | data.ZIF8_Snurr_rep222 | data.CuBTC_CaleroIV_rep111 | data.IRMOF1_mCBAC_rep111 |
Note: all three MOFs have cubic unit cells
The result of each simulation is the PNPD and average potential energy (for each N state). The PNPD may be used to compute the adsorption isotherm by the histogram-reweighting procedure described by Siderius and Shen [10]. All systems were single phase and, hence, no phase decomposition of the PNPD was necessary. The pressure for a particular chemical potential was determined from GC-TMMC simulation of the bulk N2 fluid [4]; the PNPD and average potential energy of the bulk fluid are also provided in the associated data repository.
The isosteric heat of adsorption was computed as
$$ q_{st} = k_B T -\dfrac{<UN>-<U><N>}{<N^2> - <N>^2}$$
The <...> brackets indicate grand-canonical averages
Results
Temperature = 300K
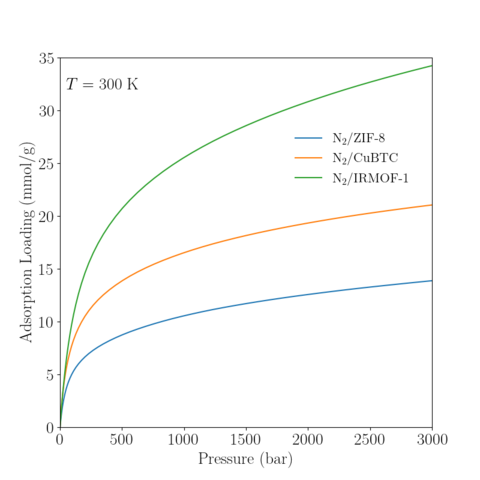
|
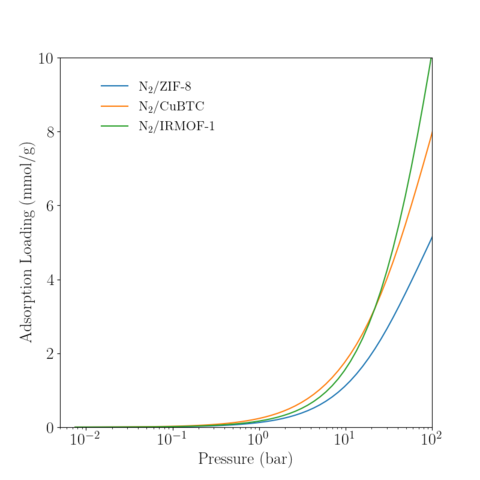
|
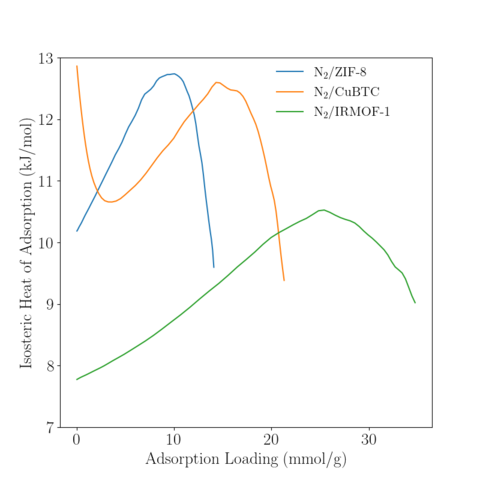
Temperature = 350 K
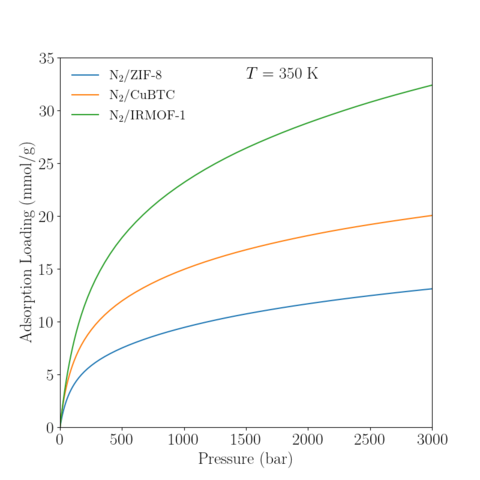
|
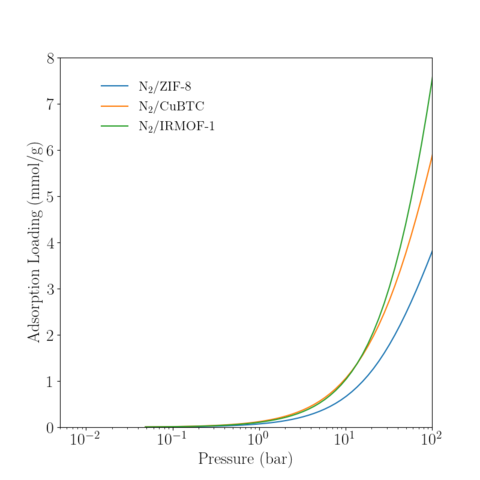
|
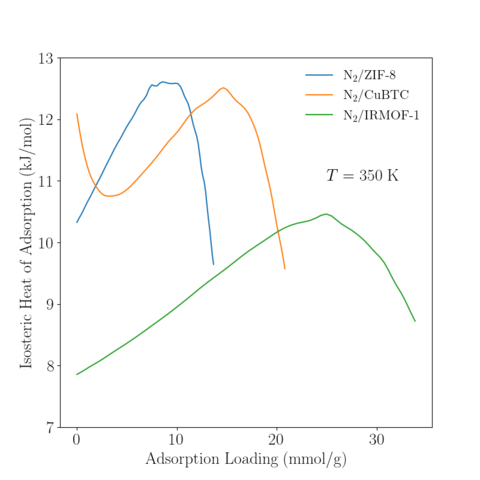
Data Availability
Various data files used to generate the reference isotherms are available in a Git Repository: https://github.com/dwsideriusNIST/NIST_SRSW_Data/tree/master/N2_REF_ISOTHERMS
Files in the repository include:
FEASST particle files for the MOF materials [includes atomic coordinates and the forcefield parameters]
FEASST particle file for TraPPE N2
Particle number probability distributions and canonical energy averages for both the bulk and adsorbed N2
Isotherm data files, including the adsorption isotherm and isosteric heat and estimated uncertainties, formatted as AIF files [21]
References
- J. R. Errington, J. Chem. Phys. 118, 9915 (2003).
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids (Oxford University Press, New York, 1989).
- D. Frenkel and B. Smit, Understanding Molecular Simulation, 2nd ed. (Academic, San Diego, 2002)., pp.37-38.
- J. R. Errington and A. Z. Panagiotopoulos, J. Chem. Phys., 109, 1093 (1998).
- J. R. Errington and V. K. Shen, J. Chem. Phys., 123, 164103 (2005).
- V. K. Shen and D. W. Siderius, J. Chem. Phys., 140, 244106, 2014.
- V. K. Shen and J. R. Errington, JPC B 108, 19595, 2004.
- V. K. Shen and J. R. Errington, JCP 122, 064508, 2005.
- V. K. Shen, R. D. Mountain, and J. R. Errington, JPC B 111, 6198, 2007.
- D. W. Siderius and V. K. Shen, JPC C 117, 5681, 2013.
- J. J. Potoff and J. I. Siepmann, AIChE J., 47, 1676–1682, 2001.
- CODATA Internationally recommended 2018 values of the Fundamental Physical Constants
- T. J. H. Vlugt, M. G. Martin, B. Smit, J. I. Siepmann, and R. Krishna, Mol Phys, 94, 727, 1998.
- H. W. Hatch, N. A. Mahynski, and V. K. Shen J Res Natl Inst Stan, 123, 123004, 2018.
- E. I. Todorov and W. Smith, The DL\_POLY User Manual (version 4.03).
- H. Zhang and R. Q. Snurr, JPC C, 121, 24000, 2017.
- J. M. Castillo, T. J. H. Vlugt, and S. Calero, JPC C 112, 15934, 2008.
- P. Ghosh, Y. J. Colón, and R. Q. Snurr, Chem Commun, 50, 11329, 2014.
- S. L. Mayo, B.D. Olafson and W. A. Goddard, JPC, 94, 8897, 1990.
- C. Zou, D. R. Penley, E. H. Cho, and L-C. Lin, JPC C 124, 11428, 2020.
- J. D. Evans, V. Bon, I. Senkovska, S. Kaskel, Langmuir, 37, 4222, 2021.