
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Understanding Protein Solution Phase Behavior via Coarse-Grained Modeling
Summary
Much of our knowledge of protein folding comes from experiments on polypeptides in dilute solutions or from theoretical models of isolated proteins. However, neither biological cells nor protein solutions encountered in biopharmaceutical development can be described as dilute. Instead, they are concentrated or "crowded" with solutes such as proteins, sugars, surfactants, salts, DNA, and fatty acids. In addition to influencing the solution thermodynamics, their presence can have a significant effect on protein denaturation, aggregation, and precipitation. To investigate these and other related questions with computer simulations requires models simple enough to allow for the efficient simulation of hundreds to thousands of protein molecules in solution, which precludes the use of all-atom descriptions of either the proteins or the solvent. We have developed multiple coarse-grained modeling strategies that account for the intrinsic free energy of folding of a protein in solvent, the main structural features of the native state and the effective protein-protein interactions. Such coarse-grained models can be used as a general tool for understanding experimental trends regarding how concentration or crowding impact the thermodynamic stability of globular proteins.
Description
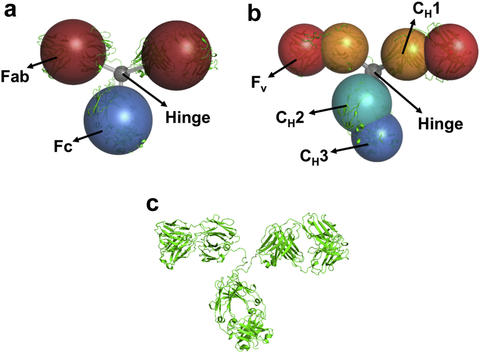
Coarse-grained models of Monoclonal antibodies with (a) 4 beads, (b) 7 beads and (c) a cartoon representation of the all-atom structure. We use computational techniques to validate the coarse-grained models and make predictions compared with small-angle scattering experiments.
Intended Impact
Because biological systems span a broad range of length and time scales, they cannot be studied effectively using conventional all-atom simulations. While hardware advances represent one route to improving the computational tractability of simulating biological systems, significant progress can be made via theory through the development of more efficient computational algorithms and simplified models describing protein solutions.
Objective
We have developed a general framework for modeling proteins in concentrated and crowded solutions. One approach accounts for both the intrinsic thermodynamics of folding and the general physical characteristics of the native and denatured states. Protein–protein interactions are derived using the salient physical features of the native and denatured conformations predicted by heteropolymer collapse theory. Another approach uses all-atom models to inform coarse-grained model interactions. Either way, we can study the effects of protein concentration and crowding on protein stability in a computationally efficient manner using transition-matrix Monte Carlo. This approach provides a general theoretical framework to study the generic effects of environmental factors (e.g., temperature, pressure, composition) on protein solution stability.
Goals
Using our coarse-grained model, we have studied the effect of temperature, concentration, macrolecular crowding, and protein sequence information on the stability of protein solutions, including aggregation behavior.
Research Activities
One general approach builds upon the predictions of heteropolymer collapse theory to provide intrinsic protein folding thermodynamics and inter-protein interactions. The result provides a reactive forcefield that can be used to simulate proteins in solution. Another approach uses all-atom structural details but assumes rigid domains. To simulate these generic systems, we use highly efficient transition-matrix Monte Carlo to calculate thermodynamic properties over a large range of state points from a single simulation.
Major Accomplishments
We first examined aqueous solutions of a single species of foldable proteins with isotropic interactions. We found that solutions of proteins with low sequence hydrophobicity are predicted to exhibit a single liquid phase over a wide range of protein concentrations and temperatures. On the other hand, solutions containing proteins with high sequence hydrophobicity display the type of temperature-inverted, first-order L-L transition that is typically associated with hydrophobic aggregation processes of amphiphilic molecules in aqueous solutions. The predicted trends for how sequence hydrophobicity modifies the relative locations of the L-L phase transition and the equilibrium unfolding curve appear to qualitatively agree with the observed solution behavior of hemoglobin HbA and its sickle variant HbS. Moreover, the results suggest that a first-order L-L transition resulting in significant protein denaturation should be expected to be found on the phase diagram of high-hydrophobicity protein solutions. The concentration fluctuations associated with such a transition could, in principle, be an important thermodynamic driving force for the nonnative aggregation that occurs below the midpoint folding temperature in solutions of high hydrophobicity proteins such as myoglobin.
We also studied the effects of anisotropic protein–protein interactions on protein stability and self-assembly behavior. In contrast with the nondirectional proteins, the strongly directional proteins we studied were stabilized against denaturation at even low protein concentrations by forming highly ordered chains. This behavior is similar to the oligomerization and polymerization of proteins in solution.
The effects of crowding agents on the conformational equilibria of proteins and thermodynamic phase behavior of their solutions have also been studied using this coarse-grained model. At low to moderate protein concentrations, crowding agents can either stabilize or destabilize the native state, depending on the strength of their attractive interaction with the proteins. At high protein concentrations, crowders tend to stabilize the native state due to excluded volume effects, irrespective of the strength of the crowder-protein attraction. Crowding agents reduce the tendency of protein solutions to undergo a liquid-liquid phase separation driven by strong protein-protein attractions. These equilibrium trends may impact how crowding species affect the driving forces for various mechanisms of physical degradation in protein solutions.
ASSOCIATED PUBLICATIONS
- Y. Xu, M. A. Blanco, M. M. Castellanos, C. W. Meuse, K. Mattison, I. Karageorgos, H. W. Hatch, V. K. Shen, J. E. Curtis, "Role of Domain-Domain Interactions on the Self-association and Physical Stability of Monoclonal Antibodies: Effect of pH and Salt", J. Phys. Chem. B, 127, 39, 8344-8357, (2023).
- Blanco, M. A., Hatch, H. W., Curtis, J. E., and Shen, V. K., "Evaluating the Effects of Hinge Flexibility on the Solution Structure of Antibodies at Concentrated Conditions," Journal of Pharmaceutical Sciences, 108, 1663-1674 (2019).
- M. A. Blanco, H. W. Hatch, J. E. Curtis, and V. K. Shen, "A methodology to calculate small-angle scattering profiles of macromolecular solutions from molecular simulations in the grand-canonical ensemble", J. Chem. Phys., 149, 084203, (2018).
- Siderius, D. W., Krekelberg, W. P., Roberts, C. J., and Shen, V. K., "Osmotic virial coefficients for model protein and colloidal solutions: Importance of ensemble constraints in the analysis of light scattering data," Journal of Chemical Physics, 136, (2012).
- V. K. Shen, J. K. Cheung, J. R. Errington, and T. M. Truskett, "Insights into crowding effects on protein stability from a coarse-grained model", Journal of Biomechanical Engineering 131, 071002 (2009).
- J. K. Cheung, V. K. Shen, J. R. Errington, and T. M. Truskett, "Concentration and crowding effects on protein stability from a coarse-grained model", in Statistical Mechanics of Cellular Systems and Processes, M. H. Zaman, ed. Cambridge University Press, In press (2009).
- J. K. Cheung, V. K. Shen, J. R. Errington, and T. M. Truskett, "Coarse-grained strategy for modeling protein stability in concentrated solutions III: Directional protein interactions", Biophysical Journal 92, 4316–4324 (2007).
- V. K. Shen, J. K. Cheung, J. R. Errington, and T. M. Truskett, "Coarse-grained strategy for modeling protein stability in concentrated solutions II: Phase behavior", Biophysical Journal 90, 1949-1960 (2006).