
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
The Inorganic Crystal Structure Database (ICSD, SRD 3), maintained at NIST, contains over 100,000 crystal structures which are known to exist and could potentially have exploitable functional properties; however, the very size of the database makes deciding which compounds to focus on challenging. To address this issue, we are using high-throughput first principles calculations to quickly screen hundreds of materials for suitability in applications, after which we can focus experimental efforts on only the most promising compounds. For example, we have used these techniques to search for new oxide thermoelectrics, finding several which may have better properties than existing materials. As a second example, we have searched for new ferroelectrics, finding many candidate materials which differ from known perovskite ferroelectrics in interesting ways.
Description
The combination of increased computing power and the improvement and streamlining of numerical techniques over the past decade have allowed for high quality first principles density functional theory calculations to be routinely performed on large numbers of compounds. This computational power, when combined with high quality materials databases, can be used to predict many materials properties without experimental input. This allows us to use high-throughput computations to screen hundreds or thousands of materials to find those few with superior properties.
Using high throughout calculations to study a given class of materials requires developing computationally efficient predictors for the properties of interest. Thus far, we have focused our efforts on performing high-throughput calculations to discover improved oxide thermoelectrics and ferroelectrics. Future work includes expanding high-throughput computations to more challenging materials and properties.
Major Accomplishments
Oxide Thermoelectrics:
Thermoelectrics are materials which can convert a temperature gradient into electric power, allowing for the generation of electricity directly from waste heat sources. Oxides have many potentially attractive qualities as thermoelectrics, including cost, stability, high effective masses, and low thermal conductivity; however, few high quality oxide thermoelectrics are known. We use a combination of high throughput calculations and modeling to develop techniques to quickly screen transition metal oxides, nitrides, and sulfides for those with high power factors and low thermal conductivity. We find several examples with predicted properties which are similar to or surpass known n-type oxide thermoelectrics.
New Ferroelectrics:
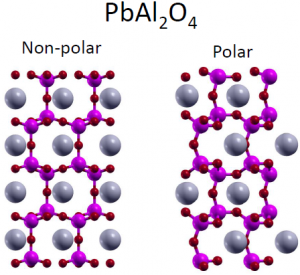
Ferroelectrics  are insulators with a non-zero polarization at low temperatures, the direction of which can be switched by an external electric field. Typical oxide ferroelectrics include distorted perovskites like BaTiO3. In this work, we search for new ferroelectrics by looking for materials with both a polar ground state and a second high-symmetry phase which is close in energy to the polar phase, which is necessary for switching. We find a variety of materials with these properties, many of which have significantly different ferroelectric mechanisms than BaTiO3. Hopefully, these new ferroelectrics will be useful in applications like memory devices, piezoelectrics, or as multiferroics.
are insulators with a non-zero polarization at low temperatures, the direction of which can be switched by an external electric field. Typical oxide ferroelectrics include distorted perovskites like BaTiO3. In this work, we search for new ferroelectrics by looking for materials with both a polar ground state and a second high-symmetry phase which is close in energy to the polar phase, which is necessary for switching. We find a variety of materials with these properties, many of which have significantly different ferroelectric mechanisms than BaTiO3. Hopefully, these new ferroelectrics will be useful in applications like memory devices, piezoelectrics, or as multiferroics.
Finite Temperature and Solid Solutions:
The vast majority of high-throughput DFT calculations have been applied to perfectly periodic undistorted crystals, but technologically relevant materials are always at a finite temperature and are often solid solutions, which creates significant computational challenges. We are working on techniques to model some of these finite temperature and doping effects in a systematic and efficient manner.