
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
Summary
As part of NIST's response to the Materials Genome Initiative (MGI), this project provides resources to address some of the challenges to the wider use of quantitative classical atomistic simulations (e.g. molecular dynamics or Monte-Carlo). This is done through:
- the development of the NIST Interatomic Potentials Repository and associated outreach activities to demonstrate that the choice of interatomic potential can significantly affect simulation results
- workshops on "Atomistic Simulations for Industrial Needs" to understand and address challenges to industrial use of these methods
- thermodynamic, structural, and kinetic property evaluations for various interatomic potentials (force fields) with comparison to available experiments and/or reliable density functional theory calculations
- the increased use of tools (e.g., IPython notebook) for reproducibility and faster evaluation of interatomic potentials
- collaboration with the wider community to facilitate the development of common formats and evaluation protocols for atomistic simulations
Description
Atomistic simulations are increasingly being used as a tool to understand and predict properties of materials in systems, such as nanomaterials, where direct measurement is time-consuming or extremely difficult. The success of atomistic simulations depends critically on the fidelity of a specific model of interatomic interactions to the area of application. Interatomic potentials in metallic systems are typically derived and optimized against a relatively small number of validating experiments and first-principles calculations, and they necessarily work best for specific materials and phases. Thus, these potentials may accurately model one system and range of parameter space but be inappropriate for another. It is therefore essential that engineers and researchers know which interatomic potentials are available and how well they reproduce material properties of interest.
To build and disseminate a repository of interatomic potentials, evaluations, and reference data, we began by soliciting feedback from researchers in industry, government, and academia to ascertain which properties are most relevant for industrial design processes. Such properties include, but are not limited to, molar volumes, phase stability, microstructural information, thermodynamics of interfaces and surfaces, diffusion, and melting temperatures. Additionally, it is important to know how these quantities change with decreasing size, especially as nanoscale materials become increasingly important. This feedback has been incorporated throughout the project, and is reflected in the activities described below.
Major Accomplishments
Among the most requested items was the development of a trusted location for the dissemination of interaction models. The NIST Interatomic Potentials Repository (www.ctcms.nist.gov/potentials/) answers this need by posting files of known origin with full citation information and notes about the formatting and use of the files in molecular simulation software. Various element and alloy interatomic potentials are available from multiple developers, and the number continues to grow. Users and developers have also requested conversions of interatomic interaction models between different file formats, and we include some potentials in multiple formats to reflect this.
Additionally, since 2008, we have hosted the annual NIST/MML Workshops on Atomistic Simulations for Industrial Needs to facilitate interactions between researchers in industry, academia, and government on issues related to the development and use of interatomic interaction potentials. Such issues include accuracy, standardization, and evaluation methods. Participants in this workshop have included representatives from the automotive, aerospace, materials design and software, and advanced materials industries. There has also been significant participation from university researchers, NIST, and various national laboratories (e.g., Sandia, Air Force Research Lab, Ames, and Los Alamos).
We have also performed structural, thermodynamic, and kinetic analyses on several popular models of aluminum optimized to have good liquid properties but which display qualitatively different crystallization behavior1. The goal of that work was to document these differences in a systematic way, and thus we examined the pair correlation functions, thermal expansions, and liquid diffusion coefficients, respectively, as functions of temperature and pressure. These results are compared with experimental information where possible. This has helped identify issues related to technical implementation of property evaluations in order to maintain consistency in approach (simulation times, data analysis methods, etc.). We find quantitative differences between the liquid properties, highlighting the importance of the chosen interatomic potential in atomistic simulations.
Besides the aluminum melt studies, we have also made a systematic comparison of stacking fault energies predicted using eight interatomic potentials for nickel2. Again we found that the choice of model could substantially affect the simulation results. We are currently examining liquid gold and nickel, as well as thermal expansions and elastic moduli of crystalline aluminum.
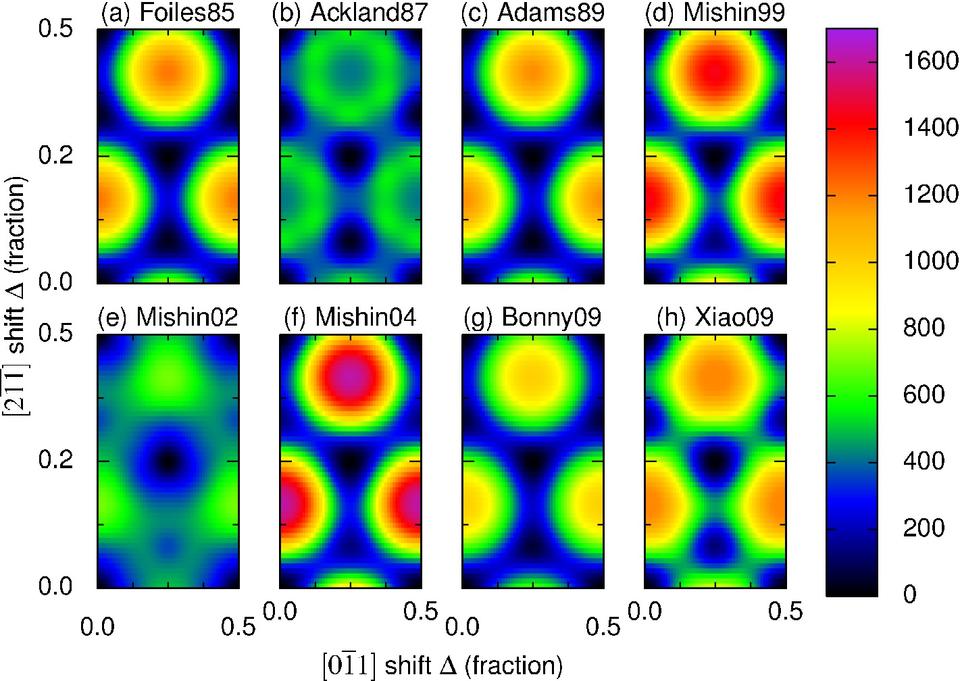
Gamma surfaces for eight interatomic potentials representing nickel2.
1. C.A. Becker and M.J. Kramer, "Atomistic comparison of volume-dependent melt properties from four models of aluminum," Modelling Simul. Mater. Sci. Eng., 18, 074001 (2010).
2. C.A. Becker, F. Tavazza, and L.E. Levine, "Implications of the choice of interatomic potential on planar faults and surface properties in nickel," Phil. Mag., 91, 3578-3597 (2011).