
Official websites use .gov
A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS
A lock (
) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
SAT-TMMC: Liquid-vapor coexistence properties - Linear-Force Shifted Potential at 2.5σ
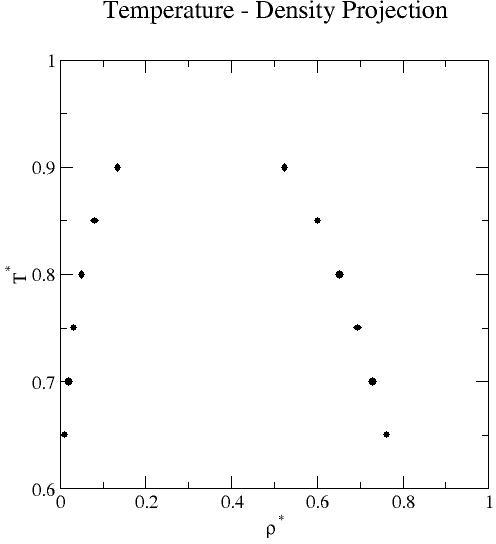
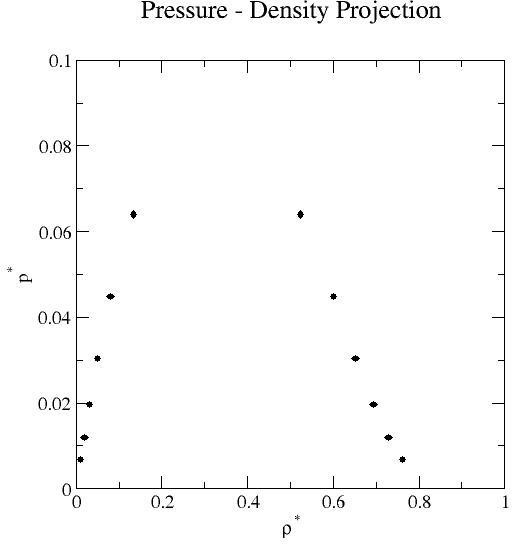
Liquid-vapor coexistence properties obtained by grand-canonical transition-matrix Monte Carlo and histogram re-weighting over the reduced temperature range 0.65 to 0.90 at increments of 0.05. Mean values of the saturation pressure, density, potential energy per molecule, and activity (chemical potential- listed below) for each phase are reported.
| METHOD | Grand-canonical transition-matrix Monte Carlo and histogram re-weighting [1, 8-12] |
| V/σ3 | 512 |
| TRUNCATION | Linear Force Shifted at 2.5σ |
| Prob. of Disp. Move | 0.4 |
| Prob. of Ins/Del Move | 0.6 |
| Biasing Function Update Frequency | 1.0E6 trial moves |
| Simulation Length | 4.0E10 trial moves |
T* | ρvap* | +/- | ρliq* | +/- | psat* | +/- | Uvap* | +/- | Uliq* | +/- | lnzsat* | +/- |
| 0.65 | 1.131E-02 | 2.386E-06 | 7.617E-01 | 1.752E-04 | 6.713E-03 | 1.280E-06 | -1.117E-01 | 2.445E-05 | -4.215E+00 | 1.084E-03 | -4.655E+00 | 1.742E-04 |
| 0.70 | 1.951E-02 | 1.673E-06 | 7.293E-01 | 1.932E-04 | 1.190E-02 | 8.675E-07 | -1.802E-01 | 1.618E-05 | -3.998E+00 | 4.682E-04 | -4.195E+00 | 6.352E-05 |
| 0.72871 | 2.560E-02 | 4.982E-06 | 7.092E-01 | 4.567E-05 | 1.594E-02 | 3.217E-06 | -2.321E-01 | 4.600E-05 | -3.868E-00 | 3.632E-04 | -3.966E+00 | 5.500E-05 |
| 0.75 | 3.188E-02 | 4.344E-06 | 6.933E-01 | 8.777E-05 | 1.954E-02 | 2.068E-06 | -2.777E-01 | 3.990E-05 | -3.767E+00 | 7.039E-04 | -3.812E+00 | 8.650E-05 |
| 0.80 | 5.044E-02 | 1.233E-05 | 6.521E-01 | 1.511E-04 | 3.025E-02 | 4.990E-06 | -4.163E-01 | 1.077E-04 | -3.515E+00 | 7.528E-04 | -3.491E+00 | 1.236E-04 |
| 0.85 | 7.951E-02 | 1.371E-05 | 6.010E-01 | 1.363E-04 | 4.475E-02 | 4.009E-06 | -6.231E-01 | 1.149E-04 | -3.225E+00 | 6.428E-04 | -3.218E+00 | 5.933E-05 |
| 0.90 | 1.350E-01 | 3.463E-05 | 5.244E-01 | 2.051E-04 | 6.398E-02 | 4.350E-06 | -1.007E+00 | 2.752E-04 | -2.843E+00 | 9.866E-04 | -2.986E+00 | 3.581E-05 |
Remarks:
Uncertainties were obtained from five independent simulations and represent 95% confidence limits based on a standard t statistic. Liquid-vapor coexistence was determined by adjusting the activity such that the pressures of the liquid and vapor phases were equal. Here, the pressure is not the conventional virial pressure [2,3] but is the actual thermodynamic pressure, based on the fact that the absolute free energies can be obtained from the distributions determined from simulation [4]. Alternative methods, for example Gibbs-ensemble Monte Carlo and combination grand-canonical Monte Carlo and histogram re-weighting, can be used to determine liquid-vapor coexistence. A review of standard methods of phase equilibria simulations can be found in Ref. 5.
As introduced in Refs. 2 and 3, the activity, z, is defined as
$$ z = \dfrac{ \exp\left( \beta \mu \right)}{\lambda^3}$$
where λ is the de Broglie wavelength, β = 1/(kBT) (where kB is Boltzmann's constant), and μ is the chemical potential. It is sometimes more convenient to work with ln z in the simulations and in post-processing. (NOTE: The reported activity is dimensionless, having been scaled by the LJ length cubed.)
Phase-coexistence energies were obtained by determining the mean potential energy at a given value of N for an additional 40 billion MC trials. Combining this information with the particle number probability distribution, the mean potential energy of the coexisting phases can be calculated [6].
For the Lennard-Jones fluid, linear force shifted at 2.5σ, the critical properties are Tc*=0.937, ρc*=0.320, and pc*=0.0820 [7].
References
- J. R. Errington, J. Chem. Phys. 118, 9915 (2003).
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids (Oxford University Press, New York, 1989).
- D. Frenkel and B. Smit, Understanding Molecular Simulation, 2nd ed. (Academic, San Diego, 2002)., pp.37-38.
- J. R. Errington and A. Z. Panagiotopoulos, J. Chem. Phys., 109, 1093 (1998).
- A. Z. Panagiotopoulos, J. Phys.: Condens. Matter, 12, R25-R52, (2000).
- J. R. Errington and V. K. Shen, J. Chem. Phys., 123, 164103 (2005).
- J. R. Errington, P. G. Debenedetti, and S. Torquato, J. Chem. Phys., 118, 2256 (2003).
- V. K. Shen and D. W. Siderius, J. Chem. Phys., 140, 244106, 2014.
- V. K. Shen and J. R. Errington, JPC B 108, 19595, 2004.
- V. K. Shen and J. R. Errington, JCP 122, 064508, 2005.
- V. K. Shen, R. D. Mountain, and J. R. Errington, JPC B 111, 6198, 2007.
- D. W. Siderius and V. K. Shen, JPC C 117, 5681, 2013.